
Abstract
Targeting the glutamatergic system has been suggested as a promising new option for developing treatment strategies for bipolar depression. Cytidine, a pyrimidine, may exert therapeutic effects through a pathway that leads to altered neuronal-glial glutamate cycling. Pyrimidines are also known to exert beneficial effects on cerebral phospholipid metabolism, catecholamine synthesis, and mitochondrial function, which have each been linked to the pathophysiology of bipolar depression. This study was aimed at determining cytidine’s efficacy in bipolar depression and at assessing the longitudinal effects of cytidine on cerebral glutamate/glutamine levels. Thirty-five patients with bipolar depression were randomly assigned to receive the mood-stabilizing drug valproate plus either cytidine or placebo for 12 weeks. Midfrontal cerebral glutamate/glutamine levels were measured using proton magnetic resonance spectroscopy before and after 2, 4, and 12 weeks of oral cytidine administration. Cytidine supplementation was associated with an earlier improvement in depressive symptoms (weeks 1–4; p=0.02, 0.001, 0.002, and 0.004, respectively) and also produced a greater reduction in cerebral glutamate/glutamine levels in patients with bipolar depression (weeks 2, 4, and 12; p=0.004, 0.004, and 0.02, respectively). Cytidine-related glutamate/glutamine decrements correlated with a reduction in depressive symptoms (p=0.001). In contrast, these relationships were not observed in the placebo add-on group. The study results suggest that cytidine supplementation of valproate is associated with an earlier treatment response in bipolar depression. Furthermore, cytidine’s efficacy in bipolar depression may be mediated by decreased levels of cerebral glutamate and/or glutamine, consistent with alterations in excitatory neurotransmission.
Similar content being viewed by others
INTRODUCTION
Bipolar disorder (BD) is a psychiatric disorder with a 1.3–1.6% lifetime prevalence and high disability (Belmaker, 2004; Muller-Oerlinghausen et al, 2002). In patients with BD, depressive episodes are more common and require a longer recovery time than manic episodes (Calabrese et al, 2004; Keller et al, 1986). Moreover, bipolar depression has been associated with a higher risk for suicide than unipolar depression (Benazzi, 2007).
Mood stabilizers and atypical antipsychotics have been established as standard treatments for bipolar mania (Muller-Oerlinghausen et al, 2002). In contrast, there has been less consensus and more controversy regarding treatment options for bipolar depression. Treatment strategies vary and include an increased dose of a mood stabilizer, the addition of a second mood stabilizer, and the adjunctive use of antidepressants (Belmaker, 2007). There remains an unmet need for developing new treatment strategies for bipolar depressed patients.
Mounting evidence supports the view that the glutamatergic system plays an important role in mediating synaptic plasticity and long-term cellular health or atrophy, factors that are presumed to be involved in the pathophysiology of BD (Sanacora et al, 2008; Zarate et al, 2006a). Modification of the glutamatergic system has been recommended as a potential treatment strategy for this disorder (Sanacora et al, 2008; Zarate et al, 2006b; Krystal et al, 2002). Recent preclinical studies indicating increased cerebral glutamate levels and reduced glia, which recycle glutamate, in BD (Dager et al, 2004; Michael et al, 2003; Ongur et al, 1998; Rajkowska, 2000; Rajkowska et al, 2001; Webster et al, 2001) also provide a plausible rationale for applying agents that modulate this system in treating patients with BD (Sanacora et al, 2008; Zarate et al, 2006a; Krystal et al, 2002).
Antidepressant effects of riluzole have been reported in patients with bipolar (Zarate et al, 2005) and unipolar depression (Zarate et al, 2004; Sanacora et al, 2007). Both riluzole and lamotrigine reduce glutamate release through mechanisms that inactivate voltage-dependent sodium channels (Ahmad et al, 2004; Lees and Leach, 1993; Benoit and Escande, 1991; Hebert et al, 1994), and recent clinical studies of both pharmaceuticals (Zarate et al, 2005; Calabrese et al, 1999) suggest that this regulation of the glutamatergic system is critical in treating bipolar depression. In accordance with the increasing need for providing therapeutics with novel mechanisms (Sanacora et al, 2008), lamotrigine has been more frequently considered as the first-line medication for the depressed phase of BD (reviewed by Fountoulakis et al, 2005).
Cytidine 5-diphosphocholine (CDP-choline), which functions in the brain as cytidine and choline (Kennedy and Weiss, 1956; Secades and Lorenzo, 2006), has received attention regarding its neuroprotective effects on a diverse range of neurological diseases (Secades and Lorenzo, 2006; Adibhatla and Hatcher, 2005; Adibhatla et al, 2002). Although precise mechanisms remain unclear, recent preclinical evidence suggests that cytidine exerts a neuroprotective effect through an increase in cerebral high-energy phosphate levels and the reversal of excess glutamate neurotransmission (Hurtado et al, 2005; Mir et al, 2003; Radad et al, 2007).
Considering that mitochondrial dysfunction has been implicated as an important element in the pathophysiology of BD (Kato and Kato, 2000; Stork and Renshaw, 2005) and that the de novo synthesis of pyrimidines (cytidine and uridine) is an energy-requiring step (Loffler et al, 2005; Santos et al, 1968), exogenous pyrimidine administration will quite likely reduce mitochondrial energy requirement in bipolar patients. Increased reservoirs of high-energy phosphate produced by cytidine might alleviate mitochondrial dysfunction and normalize energy-requiring glia-neuronal glutamatergic cycling in BD.
Cytidine, a pyrimidine, is also known to play a role in phospholipid metabolism and membrane stability (Cansev, 2006; Carlezon et al, 2005; Savci and Wurtman, 1995). Biochemical alterations in the structure and function of neuronal membranes can affect the intracellular and extracellular processes and synaptic neurotransmission, all of which may be involved in the pathophysiology of mood disorders (Carlezon et al, 2005; Lands, 1992; Nomura et al, 2001; Pacheco and Jope, 1996). Pyrimidines have also shown antidepressant-like effects in animal studies (Carlezon et al, 2002, 2005), which may stem from the alteration of cerebral neurotransmission (Carlezon et al, 2002; Martinet et al, 1978, 1979). Experimental reports that CDP-choline increases the cerebral dopamine and norepinephrine levels suggest cytidine’s effects on synaptic catecholamine transmission (Martinet et al, 1978, 1979; Agut et al, 1984; Saligaut et al, 1985).
Taken together, cytidine, which has apparent beneficial effects on cerebral energy and neuronal membrane phospholipid metabolism, may prove to be beneficial in treating patients with BD through its influence on the glutamatergic system.
In this study, we aimed to investigate whether oral cytidine supplementation is beneficial in improving depressive symptoms in bipolar depressed patients who are being treated with valproate. We also aimed to test the hypothesis, using in vivo proton magnetic resonance spectroscopy (MRS), that oral cytidine administration would induce changes in cerebral glutamate/glutamine levels.
MATERIALS AND METHODS
Subjects
Thirty-five patients who met DSM-IV criteria for bipolar I or II disorder, depressive state were recruited from the psychiatric clinic at the Catholic University College of Medicine. Image data were analyzed in collaboration with the McLean Hospital Brain Imaging Center and the Seoul National University Hospital. Inclusion criteria were (1) age 20–65 years, (2) BD I or II, as determined by the Structured Clinical Interview for DSM-IV-Patient version (SCID-P) (First et al, 2002), and (3) depressive phase (17-item Hamilton Depression Rating Score, HDRS >18) (Hamilton, 1960). Exclusion criteria included major medical, neurological disorders, comorbid psychiatric disorders (with the exception of anxiety disorders and non-antisocial/borderline personality disorders), and contraindications to magnetic resonance imaging (MRI).
The study protocol and consent form were approved by the Institutional Review Boards at the respective institutions. After a complete description of the study, written informed consent was obtained from all subjects before participation.
Patients with bipolar depression were randomly assigned to receive valproate plus either placebo or oral cytidine supplementation. Before randomization, a minimum 1-week washout period was required in patients who were on mood stabilizers or antimanic drugs other than valproate.
The treatment plan for all subjects included valproate loading and maintenance. Once a target plasma concentration (50–100 μg/ml) was achieved over a 5-day period, valproate dosing remained unchanged. Zolpidem (5–10 mg per day) for bedtime sedation and concomitant medications for stable medical conditions were permitted.
Cytidine was administered at 1 g twice a day. Placebo was formulated as an inert fructose pill. Cytidine from ICN Biomedicals was purified by Natural Pharmacia International Inc., processed under GMP conditions (Chemo Dynamics, LP, Sayreville, NJ, USA), and purified cytidine was then encapsulated by Nutra Med Inc. (Rahway, NJ, USA) into capsules containing 250 mg cytidine.
All patients were scheduled for baseline and three additional follow-up MR examinations, at weeks 2, 4, and 12, to assess the effects of adjunctive cytidine administration on cerebral metabolite changes over a 12-week treatment period.
Clinical assessments by the HDRS were conducted at baseline and weekly intervals for the first 4-week period and then at weeks 6, 8, and 12 for the remaining treatment period.
MRI/MRS Acquisition and Processing
Brain MRI was performed using a 3.0 Tesla GE whole body imaging system (GE VH/i, USA) for 18 cytidine and 17 placebo supplementation subjects. A three-dimensional spoiled gradient (3D SPGR) echo pulse sequence was used to obtain sagittal T-1 weighted images (echo time TE=14 ms, repetition time TR=5.7 ms, 256 × 256 matrix; field of view (FOV)=22 cm, flip angle=20°, number of excitation (NEX)=1, slice thickness/skip=0.7/0 mm). Axial T-2 weighted images (TE=118 ms, TR=3500 ms, 256 × 192 matrix; FOV=22 cm, flip angle=90°, NEX=3, slice thickness/skip=5/1.5 mm) and Fluid Attenuated Inversion Recovery (FLAIR) axial images (TE=145 ms, TR=9900 ms, 256 × 192 matrix; FOV=22 cm, flip angle=90°, NEX=1, slice thickness/skip=5/1.5 mm) were obtained to screen for brain structural abnormalities.
A water-suppressed, localized point-resolved spectroscopy (PRESS) pulse sequence was performed using single-voxel proton MRS obtained with a quadrature head coil. Parameters were as follows: TR/TE=2000/35 ms, phase-cycling=8, voxel of interest (VOI)=15 × 15 × 15 mm3, acquisition time=128 × 2 s, bandwidth=2500 Hz.
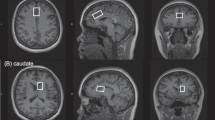
The midfrontal cortex VOI was positioned anterior to the genu of the corpus callosum, and was centered on the midline on the axial plane and on the bicommissural line on the sagittal plane, as described earlier (Ham et al, 2007; Sung et al, 2007) (Figure 1).

Voxel placement and typical in vivo spectra in the midfrontal region of interest. (a) Axial T2-weighted image showing the typical location of voxel (red box) located on the midfrontal gray matter. (b) Sample proton magnetic resonance spectra acquired from the baseline scan of one patient. LCModel estimated baselines are in smooth gray line. LCModel fit to metabolite signals are in heavy red line. The raw data is in thin gray trace. At the top of each plot, the residual signal following fitting is displayed. NAA; N-acetyl aspartate/N-acetyl aspartaryl glutamate; Glx, glutamate/glutamine; Cr, creatine+phosphocreatine; Cho, choline.
Absolute metabolite concentrations of glutamate/glutamine (Glx), N-acetyl aspartate/N-acetyl aspartaryl glutamate (NAA), creatine/phosphocreatine (Cr), and choline were estimated using the Linear Combination Model (LCModel) software (Provencher, 2001). An unsuppressed water signal was used as an internal concentration reference, and the standard GE metabolite spectra was used as a basis set (Provencher, 2001; Barker et al, 1993). Metabolite concentrations are reported as mmol/l. The macromolecule and lipid basis spectra were also included into LCModel fitting (Provencher, 2008). Spectral quality was adequate for reliable peak fitting for metabolites, with a signal-to-noise ratio (SNR) of 7.18±1.07 and a full width at half maximum (FWHM) of 0.064±0.009 p.p.m. across every time point. To ensure high-quality data, metabolite concentrations from spectra with a Cramer-Rao Lower Bound value <20% (Provencher, 2001) were included only in the final analyses. Estimates of the variances associated with metabolites other than Glx were all in the acceptable range (within 20%) in our study. Glx resonances from one 2-week follow-up scan, two 4-week follow-up scans, and one 12-week follow-up scan were excluded because of poor reliability of determination. There were no differences in gray matter, white matter, and cerebrospinal fluid (CSF) proportions at each time point between cytidine and placebo groups. Metabolite concentrations were arithmetically corrected for voxel CSF proportion assuming a metabolite concentration of zero in CSF (Bustillo et al, 2008; McLean et al, 2001).
Given the role of glutamate in the pathophysiology of BD (Krystal et al, 2002; Kugaya and Sanacora, 2005) and potential effects of cytidine on cerebral glutamate levels through mechanisms involving membrane stability and high-energy phosphate restoration (Hurtado et al, 2005), changes in cerebral glutamate/glutamine level were of interest to us.
Statistical Analysis
Group differences in demographic characteristics involving continuous and categorical data were assessed using independent t-tests and χ2 tests, respectively.
In this study, the primary outcome measure was changes in HDRS scores from baseline. The secondary outcome measure included changes in cerebral glutamate/glutamine levels. A mixed-effect regression model with repeated measures was used to analyze the primary and secondary outcomes, which included all available data at each time point. Age, sex, depression severity, and cerebral glutamate/glutamine levels at baseline were covaried when necessary. Efficacy was evaluated by testing the interaction effects of the treatment group at each time point. Response was a priori defined as a more than 50% reduction in baseline HDRS scores.
To determine the clinical relevance of glutamate/glutamine changes in each group, partial correlation analyses, controlling for age and sex composition, were performed to assess associations between changes in glutamate/glutamine levels and HDRS scores in cytidine and placebo add-on groups. On the basis of an a priori assumption that depressive symptoms and neurometabolites would change over time in a linear manner, the slope, ie, rate of changes of HDRS scores and glutamate/glutamine levels over time was used as an indicator variable.
To evaluate whether the patterns of associations between cerebral glutamate/glutamine level changes and clinical improvement during the treatment period would differ between groups, regression lines were compared, with an interaction term as an indicator, between the two groups.
Statistical significance was defined at an α-level of less than 0.05 and as two-tailed. Stata 5.0 for Windows was used for all computations.
RESULTS
Effects of Cytidine Add-On on Changes of HDRS Scores and Cerebral Glutamate/Glutamine Levels: Primary and Secondary Outcome Measures
Study subject demographics, clinical characteristics, and previously used medications are presented in Table 1. There were no significant differences in demographic or clinical characteristics between the cytidine and placebo add-on groups. Four patients (two for each group) out of 35 initially assigned patients dropped out at 3, 4, 7, and 8 weeks after baseline.
Lifetime histories of participants’ prior medication use are described in Table 1. Participants in both groups were similar regarding treatments for current episodes prior to the participation of this study. In all, 33.3, 33.3, 16.7, and 5.6% of the cytidine add-on patients received valproate, lithium, antidepressants, and neuroleptics, respectively, prior to study participation. In addition, 35.3, 29.4, 11.8, 11.8, and 5.9% of placebo add-on patients were treated for current depressive episodes with valproate, lithium, antidepressants, carbamazepine, and neuroleptics, respectively. There was no significant difference in zolpidem use between the cytidine and placebo add-on groups (χ2(1)=0.23, p=0.63).
No statistically significant difference between the two treatment groups at baseline was noted in the HDRS scores (t(33)=−0.3, p=0.77) and cerebral glutamate/glutamine levels (t(33)=−0.1, p=0.95).
The cytidine add-on group showed a significantly greater improvement in clinical symptoms than did the placebo add-on group early in the study period (Figure 2). With week-by-week mixed models, these significant differences between the two treatment groups were detectable from the first evaluation (week 1) and were maintained through week 4 (week 1, z=−2.4, p=0.02; week 2, z=−3.3, p=0.001; week 3, z=−3.1, p=0.002; week 4, z=−2.9, p=0.004) (Figure 2).

Estimated changes in HDRS scores and cerebral glutamate/glutamine levels over time. Bars represent estimated glutamate/glutamine changes from baseline in cytidine (black) and placebo (gray) add-on patients with bipolar depression. p-values above the bars indicate significant difference between treatment groups in rates of decreasing glutamate/glutamine levels throughout the treatment period with mixed-effect regression model. Squares and trend lines represent the estimated HDRS score in cytidine (black) and placebo (gray) add-on patients with bipolar depression. Asterisks represent p<0.01 significant difference between treatment groups in rates of improvement in HDRS scores from week 1–4 with mixed-effect regression model. HDRS, 17-item Hamilton Depression Rating Scale.
Response rates did not differ significantly between the cytidine add-on group and the placebo add-on group at the study end point (53.3 and 46.6%, respectively, χ2(1)=0.04, p=0.85).
The rate of changes in frontal glutamate/glutamine levels from baseline to follow-up scans in the cytidine add-on group differed significantly from that observed in the placebo add-on group throughout the treatment period (week 2, z=−2.9, p=0.004; week 4, z=−2.9, p=0.004; week 12, z=−2.3, p=0.02) (Figure 2).
There were no significant differences in the rate of changes in other cerebral metabolites including creatine, choline, or NAA between cytidine and placebo add-on groups (Supplementary Table 1).
Relationship between Rates of Change in HDRS Scores and Glutamate/Glutamine Levels
We also examined whether cerebral glutamate/glutamine changes were associated with clinical improvement in depressive symptoms in each patient group.
Cerebral glutamate/glutamine level changes were positively associated with clinical improvement in depressive symptoms (r=0.69, p=0.001) in the cytidine add-on group but not in the placebo add-on group (r=−0.34, p=0.18). Covarying for age and sex composition produced similar results (cytidine and placebo groups: r=0.67, p=0.005 and r=−0.29, p=0.29, respectively). Multivariate analysis using interaction terms has also shown that the there is a differential pattern of association between cerebral glutamate/glutamine levels and HDRS scores between the cytidine add-on patients and placebo add-on groups (p for interaction=0.009) (Figure 3).

Scatter plots and regression lines between slopes for cerebral glutamate/glutamine and HDRS changes in each treatment group. Black and gray lines represent the regression lines of change in cerebral glutamate/glutamine levels and HDRS scores in the cytidine and the placebo add-on patients, respectively. Black and gray dotted lines denote their 95% confidence intervals, respectively. Glutamate/glutamine changes were significantly associated with HDRS score changes in the cytidine add-on group (p=0.001), not in the placebo add-on group (p=0.18). Regression lines for the two groups of patients differed significantly (p=0.009). HDRS, 17-item Hamilton Depression Rating Scale.
In the cytidine add-on group, cumulative adverse events were gastrointestinal discomfort (11.1%), tremor (11.1%), headache (16.7%), restlessness (5.6%), weight gain (5.6%), and somnolence (11.1%). Adverse events of the patients assigned to the placebo add-on group included gastrointestinal discomfort (11.8%), dizziness (5.9%), headache (11.8%), somnolence (5.9%), dry mouth (5.9%), and tremor (11.8%). No serious adverse events were noted in any of the treatment groups. The profiles and frequency adverse events did not differ between the two groups.
By inclusion criteria, participating subjects did not exhibit hypomania or mania using criteria from DSM-IV and with YMRS (⩾16) at baseline. One subject from the placebo add-on group experienced treatment-emergent hypomania during the period of the study.
DISCUSSION
This double-blind, randomized, placebo-controlled study suggests that cytidine augmentation of valproate produced a much earlier improvement in depressive symptoms in patients with bipolar depression. Cytidine supplementation was also associated with greater and more rapid reductions in cerebral glutamate/glutamine levels. Although glutamate in the brain can be non-invasively measured in vivo by using proton MRS, it should be pointed out that the glutamate signal, which constitutes a major part of glutamate/glutamine resonance in the LCModel (60–80%) (Pouwels and Frahm, 1998), is hard to resolve separately from the glutamine signal even at higher magnetic fields, such as in the 3 Tesla scanner used in this study. The glutamate/glutamine resonance, therefore, has been assumed to reflect alterations in the glutamate/glutamine cycle (Rothman et al, 2003).
It is noted that decreases in cerebral glutamate/glutamine levels were associated with clinical improvement only in the cytidine add-on group. Comparison of regression lines between groups (p for slope difference=0.009, Figure 3) also showed that the cerebral Glx level changes in the cytidine group are relevant to cytidine’s therapeutic efficacy.
There was no change in cerebral glutamate/glutamine levels in the placebo add-on group. This suggests that valproate-induced clinical improvement in bipolar depression is likely to have a mechanism of action different from that in the cytidine add-on administration. Our previous proton magnetic resonance spectroscopic imaging study reported that valproate-treated bipolar patients did not show cerebral Glx level changes despite symptom improvement (Friedman et al, 2004).
Although antidepressants are frequently used in treating patients with bipolar depression (Belmaker, 2007), there have been debates about their use because of the risk of iatrogenic episodes of mania or rapid cycling as well as concerns about their efficacy (Belmaker, 2004; Gijsman et al, 2004; Moller et al, 2006). Reviews of well-controlled studies have suggested that antidepressants are efficient in the short-term treatment of bipolar depression with a relatively low risk for mood switch (Gijsman et al, 2004; Moller et al, 2006). However, a recent large-scale controlled study, conducted in accord with routine clinical practice, reported that standard adjunctive antidepressants were neither efficacious in the treatment of bipolar depression and nor did they increase the risk for mood switch (Sachs et al, 2007). These recent efficacy data highlight the need for developing new treatment strategies for bipolar depression.
A lag in treatment response, approximately 4–6 weeks, for conventional antidepressants (Hyman and Nestler, 1996) poses another clinical challenge, given the high risk for suicidal behaviors within the first 9 days after starting antidepressants (Jick et al, 2004). Therefore, treatment strategies with a more rapid onset of antidepressant effects are important in treating patients with bipolar depression.
Fluoxetine add-on to olanzapine in treating bipolar depression produced a greater improvement in depressive symptoms starting at 4 weeks to week 8 relative to the olanzapine monotherapy group (Tohen et al, 2003). Considering the current observation of faster, as early as week 1, clinical improvement, cytidine's mechanism for re-establishing normal mood in bipolar depression may be different from those of antidepressants.
Adaptive neurotrophic signaling changes have been postulated as a final common pathway for stable long-term treatment effects of mood disorders (Kugaya and Sanacora, 2005). A focus on glutamatergic system-associated synaptic plasticity has specifically been proposed as one mechanism involved in the onset of antidepressant actions (Sanacora et al, 2008; Machado-Vieira et al, 2008). Recent clinical trials reporting the efficacy of an N-methyl-D-aspartate antagonist on treatment-resistant depression supports the role of the glutamatergic system in rapid and sustained treatment responses for mood disorder (Zarate et al, 2006b).
Increased cerebral glutamate levels have consistently been reported in patients with bipolar depression (Dager et al, 2004; Michael et al, 2003; Stork and Renshaw, 2005). In view of the mitochondrial dysfunction (Kato and Kato, 2000; Stork and Renshaw, 2005) and glial-cell pathology observed in BD (Ongur et al, 1998; Rajkowska, 2000; Rajkowska et al, 2001; Webster et al, 2001), high cerebral glutamate levels may potentially be due to deficits in highly energy-requiring transport of the excitatory neurotransmitter from the synaptic spaces into astrocytes. By alleviating the demand on neuronal energy metabolism, exogenous cytidine administration may help remove excess glutamate, thus preventing it from rising to neurotoxic values (Maragakis and Rothstein, 2001; Takahashi et al, 1997). Bipolar depressed patients treated with triacetyluridine, which is converted to free uridine on absorption, manifested an early treatment onset with improvement of mitochondrial function (Jensen et al, 2008). Uridine serves as the primary plasma form of pyrimidines, and cytidine and uridine interconvert (Cansev, 2006). This clinical evidence is consistent with a role for cytidine in normalizing cerebral energy metabolism in BD patients.
Exogenous cytidine administration may also produce an increased synthesis of phospholipids in brain cell membranes (Loffler et al, 2005; Cansev, 2006). Restoration of neuronal membrane stability could alter various processes to normalize neurotransmitter systems that may be involved in the pathophysiology of mood disorder (Carlezon et al, 2005; Nomura et al, 2001; Pacheco and Jope, 1996). CDP-choline exerts effects to increase cerebral dopamine and norepinephrine levels (Martinet et al, 1978, 1979; Agut et al, 1984; Saligaut et al, 1985). Specifically, pyrimidine-induced increase in ATP levels (Silveri et al, 2008) may enhance tyrosine hydroxylase activity (Martinet et al, 1978, 1979; Saligaut et al, 1985), an ATP-dependent and rate-limiting enzyme for dopamine and norepinephrine synthesis. Similar effects on glutamate synthesis or turnover have not been studied.
Our findings support the overall safety of cytidine supplementation of mood stabilizers in treating BD. Although the safety of short-term and long-term uses of CDP-choline, which consists of cytidine and choline, has been reported consistently (Secades and Lorenzo, 2006), the long-term tolerability of the use of cytidine in patients with BD should be evaluated in a larger cohort over a longer follow-up period.
Recently, two randomized controlled trials suggested that divalproex is likely to be an effective treatment on bipolar depression (Ghaemi et al, 2007; Davis et al, 2005). Current valproate administration in the placebo group showed a comparable efficacy at 6- and 8-week treatment periods to those in earlier studies that assessed the efficacy of valproate monotherapy (Ghaemi et al, 2007; Davis et al, 2005). Our findings suggest that cytidine add-on treatment has a beneficial effect on earlier response than valproate-alone treatment. To further support the earlier response in the cytidine add-on group, we compared the time to first response (defined as the first time there was a more than 50% reduction in baseline HDRS scores) between groups. The mean time to first response was significantly shorter in the cytidine add-on group relative to the placebo add-on group (mean time to response: 36.4 and 50.9 days, respectively; t(24)=−2.63, p=0.015).
Several factors should be taken into consideration in interpreting these data. First, considering that BD is heterogeneous in clinical characteristics and genetic determination (Muller-Oerlinghausen et al, 2002), the findings from our cohort may not be generalized. Second, without antidepressant or antidepressant plus valproate as active control arms, our study could not evaluate the relative effect size of added cytidine to antidepressant supplementation. However, considering the previously reported limited efficacy of antidepressants relative to placebo (Sachs et al, 2007), our placebo-controlled study design may provide clinically meaningful implications for treating bipolar depression.
To the best of our knowledge, this study appears to be the first report regarding the association between exogenous cytidine administration and cerebral glutamate/glutamine levels. As preclinical studies suggest that CDP-choline, which is composed of cytidine and choline, may exert a neuroprotective effect by inhibiting the glutamate-induced apoptosis (Hurtado et al, 2005; Mir et al, 2003; Radad et al, 2007), further studies in healthy subjects are warranted to confirm the physiological relationship between cytidine administration and alterations in the cerebral glutamatergic system.
In conclusion, we report that cytidine supplementation of valproate is associated with an earlier response in treating bipolar depression and with greater and earlier reductions in cerebral glutamate/glutamine levels. In light of the need for developing improved therapeutics targeting the symptoms of bipolar depression, cytidine augmentation, which may alter the glutamatergic system potentially through a novel mechanism, appears to be a promising option for further study for treating severe depressive episodes in bipolar patients.
References
Adibhatla RM, Hatcher JF (2005). Cytidine 5′-diphosphocholine (CDP-choline) in stroke and other CNS disorders. Neurochem Res 30: 15–23.
Adibhatla RM, Hatcher JF, Dempsey RJ (2002). Citicoline: neuroprotective mechanisms in cerebral ischemia. J Neurochem 80: 12–23.
Agut J, Coviella IL, Wurtman RJ (1984). Cytidine(5′)diphosphocholine enhances the ability of haloperidol to increase dopamine metabolites in the striatum of the rat and to diminish stereotyped behavior induced by apomorphine. Neuropharmacology 23: 1403–1406.
Ahmad S, Fowler LJ, Whitton PS (2004). Effects of acute and chronic lamotrigine treatment on basal and stimulated extracellular amino acids in the hippocampus of freely moving rats. Brain Res 1029: 41–47.
Barker PB, Soher BJ, Blackband SJ, Chatham JC, Mathews VP, Bryan RN (1993). Quantitation of proton NMR spectra of the human brain using tissue water as an internal concentration reference. NMR Biomed 6: 89–94.
Belmaker RH (2004). Bipolar disorder. N Engl J Med 351: 476–486.
Belmaker RH (2007). Treatment of bipolar depression. N Engl J Med 356: 1771–1773.
Benazzi F (2007). Bipolar disorder—focus on bipolar II disorder and mixed depression. Lancet 369: 935–945.
Benoit E, Escande D (1991). Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflugers Arch 419: 603–609.
Bustillo JR, Rowland LM, Jung R, Brooks WM, Qualls C, Hammond R et al (2008). Proton magnetic resonance spectroscopy during initial treatment with antipsychotic medication in schizophrenia. Neuropsychopharmacology 33: 2456–2466.
Calabrese JR, Bowden CL, Sachs GS, Ascher JA, Monaghan E, Rudd GD (1999). A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry 60: 79–88.
Calabrese JR, Hirschfeld RM, Frye MA, Reed ML (2004). Impact of depressive symptoms compared with manic symptoms in bipolar disorder: results of a U.S. community-based sample. J Clin Psychiatry 65: 1499–1504.
Cansev M (2006). Uridine and cytidine in the brain: their transport and utilization. Brain Res Rev 52: 389–397.
Carlezon Jr WA, Mague SD, Parow AM, Stoll AL, Cohen BM, Renshaw PF (2005). Antidepressant-like effects of uridine and omega-3 fatty acids are potentiated by combined treatment in rats. Biol Psychiatry 57: 343–350.
Carlezon WA, Pliakas AM, Parow AM, Detke MJ, Cohen BM, Renshaw PF (2002). Antidepressant-like effects of cytidine in the forced swim test in rats. Biol Psychiatry 51: 882–889.
Dager SR, Friedman SD, Parow A, Demopulos C, Stoll AL, Lyoo IK et al (2004). Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry 61: 450–458.
Davis LL, Bartolucci A, Petty F (2005). Divalproex in the treatment of bipolar depression: a placebo-controlled study. J Affect Disord 85: 259–266.
First MB, Spitzer RL, Gibbon M, Williams JBW (2002). Structural Clinical Interview for DSM-IV-TR, Research Version, Non-patient Edition. Biometrics Research, New York State Psychiatric Institute: New York.
Fountoulakis KN, Vieta E, Sanchez-Moreno J, Kaprinis SG, Goikolea JM, Kaprinis GS (2005). Treatment guidelines for bipolar disorder: a critical review. J Affect Disord 86: 1–10.
Friedman SD, Dager SR, Parow A, Hirashima F, Demopulos C, Stoll AL et al (2004). Lithium and valproic acid treatment effects on brain chemistry in bipolar disorder. Biol Psychiatry 56: 340–348.
Ghaemi SN, Gilmer WS, Goldberg JF, Zablotsky B, Kemp DE, Kelley ME et al (2007). Divalproex in the treatment of acute bipolar depression: a preliminary double-blind, randomized, placebo-controlled pilot study. J Clin Psychiatry 68: 1840–1844.
Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM (2004). Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 161: 1537–1547.
Ham BJ, Chey J, Yoon SJ, Sung Y, Jeong DU, Ju Kim S et al (2007). Decreased N-acetyl-aspartate levels in anterior cingulate and hippocampus in subjects with post-traumatic stress disorder: a proton magnetic resonance spectroscopy study. Eur J Neurosci 25: 324–329.
Hamilton M (1960). A rating scale for depression. J Neurol Neurosurg Psychiatry 23: 56–62.
Hebert T, Drapeau P, Pradier L, Dunn RJ (1994). Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol 45: 1055–1060.
Hurtado O, Moro MA, Cardenas A, Sanchez V, Fernandez-Tome P, Leza JC et al (2005). Neuroprotection afforded by prior citicoline administration in experimental brain ischemia: effects on glutamate transport. Neurobiol Dis 18: 336–345.
Hyman SE, Nestler EJ (1996). Initiation and adaptation: a paradigm for understanding psychotropic drug action. Am J Psychiatry 153: 151–162.
Jensen JE, Daniels M, Haws C, Bolo NR, Lyoo IK, Yoon SJ et al (2008). Triacetyluridine (TAU) decreases depressive symptoms and increases brain pH in bipolar patients. Exp Clin Psychopharmacol 16: 199–206.
Jick H, Kaye JA, Jick SS (2004). Antidepressants and the risk of suicidal behaviors. JAMA 292: 338–343.
Kato T, Kato N (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord 2: 180–190.
Keller MB, Lavori PW, Coryell W, Andreasen NC, Endicott J, Clayton PJ et al (1986). Differential outcome of pure manic, mixed/cycling, and pure depressive episodes in patients with bipolar illness. JAMA 255: 3138–3142.
Kennedy EP, Weiss SB (1956). The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem 222: 193–214.
Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G et al (2002). Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry 7 (Suppl 1): S71–S80.
Kugaya A, Sanacora G (2005). Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr 10: 808–819.
Lands WE (1992). Biochemistry and physiology of n-3 fatty acids. FASEB J 6: 2530–2536.
Lees G, Leach MJ (1993). Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neurological cultures from rat cortex. Brain Res 612: 190–199.
Loffler M, Fairbanks LD, Zameitat E, Marinaki AM, Simmonds HA (2005). Pyrimidine pathways in health and disease. Trends Mol Med 11: 430–437.
Machado-Vieira R, Salvadore G, Luckenbaugh DA, Manji HK, Zarate CA (2008). Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J Clin Psychiatry 69: 946–958.
Maragakis NJ, Rothstein JD (2001). Glutamate transporters in neurologic disease. Arch Neurol 58: 365–370.
Martinet M, Fonlupt P, Pacheco H (1978). Interaction of CDP-choline with synaptosomal transport of biogenic amines and their precursors in vitro and in vivo in the rat corpus striatum. Experientia 34: 1197–1199.
Martinet M, Fonlupt P, Pacheco H (1979). Effects of cytidine-5′ diphosphocholine on norepinephrine, dopamine and serotonin synthesis in various regions of the rat brain. Arch Int Pharmacodyn Ther 239: 52–61.
McLean MA, Woermann FG, Simister RJ, Barker GJ, Duncan JS (2001). In vivo short echo time 1H-magnetic resonance spectroscopic imaging (MRSI) of the temporal lobes. Neuroimage 14: 501–509.
Michael N, Erfurth A, Ohrmann P, Gossling M, Arolt V, Heindel W et al (2003). Acute mania is accompanied by elevated glutamate/glutamine levels within the left dorsolateral prefrontal cortex. Psychopharmacology (Berl) 168: 344–346.
Mir C, Clotet J, Aledo R, Durany N, Argemi J, Lozano R et al (2003). CDP-choline prevents glutamate-mediated cell death in cerebellar granule neurons. J Mol Neurosci 20: 53–60.
Moller HJ, Grunze H, Broich K (2006). Do recent efficacy data on the drug treatment of acute bipolar depression support the position that drugs other than antidepressants are the treatment of choice? A conceptual review. Eur Arch Psychiatry Clin Neurosci 256: 1–16.
Muller-Oerlinghausen B, Berghofer A, Bauer M (2002). Bipolar disorder. Lancet 359: 241–247.
Nomura T, Nishizaki T, Enomoto T, Itoh H (2001). A long-lasting facilitation of hippocampal neurotransmission via a phospholipase A2 signaling pathway. Life Sci 68: 2885–2891.
Ongur D, Drevets WC, Price JL (1998). Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA 95: 13290–13295.
Pacheco MA, Jope RS (1996). Phosphoinositide signaling in human brain. Prog Neurobiol 50: 255–273.
Pouwels PJ, Frahm J (1998). Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med 39: 53–60.
Provencher SW (2001). Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed 14: 260–264.
Provencher SW (2008). LCModel User’s Manual, version 6.2.
Radad K, Gille G, Xiaojing J, Durany N, Rausch WD (2007). CDP-choline reduces dopaminergic cell loss induced by MPP(+) and glutamate in primary mesencephalic cell culture. Int J Neurosci 117: 985–998.
Rajkowska G (2000). Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 48: 766–777.
Rajkowska G, Halaris A, Selemon LD (2001). Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry 49: 741–752.
Rothman DL, Behar KL, Hyder F, Shulman RG (2003). In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol 65: 401–427.
Sachs GS, Nierenberg AA, Calabrese JR, Marangell LB, Wisniewski SR, Gyulai L et al (2007). Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med 356: 1711–1722.
Saligaut C, Daoust M, Chadelaud M, Moore N, Chretien P, Boismare F (1985). Oxotremorine-induced cholinergic syndrome: modifications by levodopa and/or oral cytidine diphosphocholine. Methods Find Exp Clin Pharmacol 7: 5–8.
Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V et al (2007). Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biol Psychiatry 61: 822–825.
Sanacora G, Zarate CA, Krystal JH, Manji HK (2008). Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7: 426–437.
Santos JN, Hempstead KW, Kopp LE, Miech RP (1968). Nucleotide metabolism in rat brain. J Neurochem 15: 367–376.
Savci V, Wurtman RJ (1995). Effect of cytidine on membrane phospholipid synthesis in rat striatal slices. J Neurochem 64: 378–384.
Secades JJ, Lorenzo JL (2006). Citicoline: pharmacological and clinical review, 2006 update. Methods Find Exp Clin Pharmacol 28 (Suppl B): 1–56.
Silveri MM, Dikan J, Jensen JE, Kamiya T, Kawada Y, Renshaw PF et al (2008). Citicoline enhances frontal lobe bioenergetics as measured by phosphorus magnetic resonance spectroscopy. NMR Biomed 21: 1066–1075.
Stork C, Renshaw PF (2005). Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry 10: 900–919.
Sung YH, Cho SC, Hwang J, Kim SJ, Kim H, Bae S et al (2007). Relationship between N-acetyl-aspartate in gray and white matter of abstinent methamphetamine abusers and their history of drug abuse: a proton magnetic resonance spectroscopy study. Drug Alcohol Depend 88: 28–35.
Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, Attwell D (1997). The role of glutamate transporters in glutamate homeostasis in the brain. J Exp Biol 200: 401–409.
Tohen M, Vieta E, Calabrese J, Ketter TA, Sachs G, Bowden C et al (2003). Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 60: 1079–1088.
Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH (2001). Immunohistochemical localization of phosphorylated glial fibrillary acidic protein in the prefrontal cortex and hippocampus from patients with schizophrenia, bipolar disorder, and depression. Brain Behav Immun 15: 388–400.
Zarate Jr CA, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D et al (2004). An open-label trial of riluzole in patients with treatment-resistant major depression. Am J Psychiatry 161: 171–174.
Zarate Jr CA, Quiroz JA, Singh JB, Denicoff KD, De Jesus G, Luckenbaugh DA et al (2005). An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry 57: 430–432.
Zarate Jr CA, Singh J, Manji HK (2006a). Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry 59: 1006–1020.
Zarate Jr CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA et al (2006b). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63: 856–864.
Acknowledgements
This study was supported in part by grant from the Stanley medical Research Institute (Drs. Lyoo, Cohen, and Renshaw), from NIMH (MH58681, Dr. Renshaw), the Korean Ministry of Science and Technology (M103KV010020-08K2201-02010, Dr. Lyoo), and the Korea Research Foundation Grant funded by the Korean Government (MOEHRD, KRF-2006-331-E00187, Dr. Yoon).
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
Dr Renshaw is a consultant to GSK, Novartis, Roche, and Kyowa Hakko. He has received research support from Eli Lilly and Siemens. Drs Cohen and Renshaw are inventors on a patent application describing the use of cytidine as a treatment for BD. This application has been assigned to McLean Hospital and has not been licensed. Neither Dr Renshaw nor Dr Cohen participated in the clinical evaluation of subjects for this study. Dr Lyoo has received research support from Eli Lilly, AstraZeneca, GSK, and Organon. He has no other potential conflicts of interest to disclosure. Drs Yoon and Kim have no conflict of interest to declare.
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary information
Rights and permissions
About this article
Cite this article
Yoon, S., Lyoo, I., Haws, C. et al. Decreased Glutamate/Glutamine Levels May Mediate Cytidine’s Efficacy in Treating Bipolar Depression: A Longitudinal Proton Magnetic Resonance Spectroscopy Study. Neuropsychopharmacol 34, 1810–1818 (2009). https://doi.org/10.1038/npp.2009.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2009.2
Keywords
This article is cited by
-
Low corticosterone levels attenuate late life depression and enhance glutamatergic neurotransmission in female rats
Acta Pharmacologica Sinica (2021)
-
Assessment of glutamatergic synaptic transmission and plasticity in brain slices: relevance to bioelectronic approaches
Bioelectronic Medicine (2019)
-
Altered gut metabolome contributes to depression-like behaviors in rats exposed to chronic unpredictable mild stress
Translational Psychiatry (2019)
-
Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression
Molecular Psychiatry (2016)
-
Pharmacological Treatment of Bipolar Depression: Qualitative Systematic Review of Double-Blind Randomized Clinical Trials
Psychiatric Quarterly (2012)
