
Abstract
The neural pathways through which substance P (SP) influences fear and anxiety are poorly understood. However, the amygdala, a brain area repeatedly implicated in fear and anxiety processes, is known to contain large numbers of SP-containing neurons and SP receptors. Several studies have implicated SP neurotransmission within the amygdala in anxiety processes. In the present study, we evaluated the effects of site-specific infusions of an SP receptor antagonist, GR 82334, on conditioned fear responses using the fear-potentiated startle paradigm. GR 82334 infusion into the basolateral (BLA) or the medial (MeA) nuclei of the amygdala, but not into the central nucleus of the amygdala (CeA), dose dependently reduced fear-potentiated startle. Similar effects were obtained with GR 82334 infusion into the ventromedial nucleus of the hypothalamus (VMH), to which the MeA projects, and into the rostral dorsolateral periaqueductal gray (PAG), to which the VMH projects, but not into the deep layers of the superior colliculus/deep mesencephalic nucleus (dSC/DpMe), an output of the CeA previously shown to be important for fear-potentiated startle. Consistent with previous findings, infusion of the AMPA receptor antagonist, NBQX, into the dSC/DpMe, but not into the PAG, did disrupt fear-potentiated startle. These findings suggest that multiple outputs from the amygdala play a critical role in fear-potentiated startle and that SP plays a critical, probably modulatory role, in the MeA to VMH to PAG to the startle pathway based on these and data from others.
Similar content being viewed by others

INTRODUCTION
A large number of studies have implicated Substance P (SP) in fear and anxiety based on the ability of local infusion of SP into various parts of the brain to elicit defensive and anxiety-like effects and that SP antagonists can have anxiolytic effects (compared with Rosenkranz, 2007). SP appears to influence affective behaviors, at least in part, by actions within the amygdala (Boyce et al, 2001; Ebner et al, 2004; Kramer et al, 1998; Smith et al, 1999). Importantly, however, SP-containing neurons and SP receptors are heterogeneously distributed within this area. Neuronal immunoreactivity for SP is intense in the medial nucleus of the amygdala (MeA), scarce in the central nucleus of the amygdala (CeA), and undetectable in the basolateral nucleus of the amygdala (BLA; Castellano and McGaugh, 1989; Damalama and Swann, 1993; Emson et al, 1978; Malsbury and McKay, 1989; Ribeiro-da-Silva and Hokfelt, 2000; Roberts et al, 1982). Although the medial amygdaloid nucleus displays one of the heaviest concentrations of SP-IR terminals in the CNS, most of this immunoreactivity is of local origin … as demonstrated by local knife cuts (Emson et al, 1978) (p 265, Ribeiro-da-Silva and Hokfelt, 2000). In contrast, SP receptor immunoreactivity is moderately dense in both the MeA and the CeA (Mantyh, 2002; Boyce et al, 2001; Saffroy et al, 1988) and less intense in the BLA (Levita et al, 2003; Saffroy et al, 1988; Smith et al, 1999). These expression patterns suggest a differential involvement of amygdaloid nuclei in SP-mediated affective behaviors. In fact, increases in SP mRNA and SP release induced by emotional stressors have been found in the MeA, but not in the CeA (Ebner et al, 2004; Sergeyev et al, 2005), and an increase in SP receptor internalization, a marker of SP release, was found in the BLA (Kramer et al, 1998; Smith et al, 1999). Direct infusion of SP into the MeA was shown to be anxiogenic in the elevated plus maze test (Ebner et al, 2004), and infusion of an SP receptor antagonist into the BLA blocked maternal separation-induced vocalization (Boyce et al, 2001). These data suggest that the MeA and the BLA, but perhaps not the CeA, are important neural substrates through which SP modulates or mediates affective behaviors. However, because of differences in methodology between these studies and limited data for the CeA, the current paper examined the contribution of SP neurotransmission in each of these areas by infusing the SP receptor antagonist GR 82334 into the BLA, the MeA, or the CeA just prior to testing in the fear-potentiated startle paradigm.
We also investigated the possible neural substrates involved with SP's role in fear-potentiated startle outside the amygdala. Fear-potentiated startle is mediated by the CeA and its projection to the deep layers of the superior colliculus/deep mesencephalic nucleus (dSC/DpMe) of the rostral midbrain and, subsequently, to the primary startle reflex circuit in the brain stem (Davis et al, 1993, 1997; Hitchcock and Davis, 1991; Meloni and Davis, 1999; Rosen et al, 1991; Frankland and Yeomans, 1995; Zhao and Davis, 2004). In addition, we recently reported that fear-potentiated startle is also blocked by infusion of the AMPA receptor antagonist NBQX into the MeA (Walker et al, 2005). It is unclear how the MeA influences fear-potentiated startle as it does not project directly to the startle reflex circuit, the dSC/DpMe, or the CeA (Canteras et al, 1995). However, the MeA does have heavy projections to the ventromedial nucleus of the hypothalamus (VMH), and this pathway was found to contain SP (Canteras et al, 1994, 1995; Han et al, 1996; Shaikh et al, 1993). Lesions of the VMH have been shown to disrupt other affective behaviors (Colpaert and Wiepkema, 1976; de Oliveira et al, 1997; Dielenberg et al, 2001; Han et al, 1996; Sudakov, 1987). Moreover, SP-containing afferents from the VMH project to the periaqueductal gray (PAG—Dornan et al, 1990), which in turn projects to the pontine startle reflex circuit (Cameron et al, 1995; Canteras et al, 1994; Fendt et al, 1994; Meloni and Davis, 1999; Siegel et al, 1997). Thus, it is conceivable that the SP-containing MeA → VMH → PAG → pontine circuit may participate in or be required for the full expression of fear-potentiated startle. To evaluate this possibility, we also infused GR 82334 into the VMH, the PAG, and also the dSC/DpMe, prior to fear-potentiated startle testing.
MATERIALS AND METHODS
Subjects
Male Sprague–Dawley rats (Charles River, Raleigh, NC), weighing 400–450 g at the time of surgery, were used in this study. Animals were housed in groups of two in plastic cages and maintained on a 12-h light–dark cycle (light 0800–2000 hours) with food and water available ad libitum. We affirm that the original studies reported in this paper have been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Cannula Implantation
Before the behavioral experiments, rats were anesthetized with Nembutal (sodium pentobarbital, 50 mg/kg i.p.; Emory University School of Medicine Pharmacy, Atlanta, GA) and placed in a Kopf stereotaxic instrument (Model 900; David Kopf, Tujunga, CA) with blunt ear bars. The skull was exposed and stainless steel guide cannulas (22 gauge; Plastics One, Roanoke, VA), which were occluded with an internal dummy stylet extending 1 mm beyond the guide cannula tip, were bilaterally lowered into the brain aiming at the MeA (AP=−2.8 mm, ML=±3.5 mm, and DV=−8.5 mm), the CeA (AP=−2.6 mm, ML=±4.2 mm, and DV=−8.5 mm), the BLA (AP=−2.8 mm, ML=±5.1 mm, and DV=−8.4 mm), the VMH (AP=−3.2 mm, ML=±0.7 mm, and DV=−9.4 mm), the dorsomedial nucleus of the hypothalamus (DMH; AP=−3.2 mm, ML=±0.7 mm, and DV=−8.4 mm), the rostral dorsolateral PAG (rostral dlPAG; AP=−6.4 mm, ML=±0.5 mm, and DV=−5.0 mm), the caudal ventrolateral PAG (caudal vlPAG; AP=−7.7 mm, ML=±0.6 mm, and DV=−6.0 mm), and the dSC/DpMe (AP=−6.4 mm, ML=±1.5 mm, and DV=−5.1 mm). The AP and ML positions were measured with respect to bregma and midline, respectively, and the DV position was measured with respect to dura above the implantation site with the exception of the CeA and the BLA for which the DV position was measured with respect to the surface of the skull at bregma. The cannulas were anchored with dental cement to four jeweler screws that were previously attached to the skull. Animals were allowed to recover from the surgery for at least 7 days prior to behavioral procedures.
Startle Apparatus
Rats were trained and tested in four identical stabilimeters located in separate chambers (90 × 70 × 70 cm) that were dark, ventilated, and sound attenuated. Each stabilimeter consisted of an 8 × 15 × 15-cm Plexiglas and wire mesh cage, and was suspended within a steel frame by four compression springs. The floor of each cage consisted of four stainless steel bars (6 mm in diameter and 18 mm apart), through which footshocks were delivered. An accelerometer (PCM Piezotronics, Depew, NY) was affixed to the bottom of the cage to detect any movement of the cage. The displacement of the accelerometer caused by cage movement generated a voltage that was integrated and proportional to the velocity of cage movement. The analog output of the accelerometer was amplified (PCB, Piezotronics, Model 483B21) and digitized on a scale of 0–2500 units by an InstruNET device (Model 100B; GW Instruments, Somerville, MA) interfaced to a Macintosh G3 computer. The amplitude of startle response was defined as the maximal peak-to-peak voltage that occurred during the first 200-ms after onset of the startle-eliciting stimulus. The 200-ms sampling window allows the highest peak-to-peak movement of the cage to be accurately sampled (Cassella and Davis, 1986). A surveillance camera (Burle, Model TC 651B with a TC 9907a lens; Operational Security Systems, Atlanta, GA) was positioned behind each stabilimeter and connected to a TV monitor located outside the chamber. Background illumination was provided by a red incandescent bulb (7.5 W) located 11 cm above the stabilimeter.
A high-frequency speaker (Radio Shack Super-tweeters, range 5–40 kHz) was positioned 5 cm from the front of each stabilimeter. Constant background white noise (60 dB) was generated by a General Radio noise generator (ACO Pacific Inc., Belmont, CA) and delivered through the speaker. The startle-eliciting stimulus was a 50-ms white noise burst (5 ms rise–decay) that was generated by the Macintosh G3 computer (0–22 kHz) and delivered through the same speaker to evoke a startle response. The background noise and the white noise burst were amplified by a Radio Shack amplifier (100 W, Model MPA-200). The intensity of the sounds was frequently calibrated with a sound-level meter (Model 2235; Bruel & Kjaer, Marlborough, MA) at a distance that approximated the distance of the rat's ear to the speaker.
The visual conditioned stimulus (CS) was a 3.7-s light (70 lux) produced by an 8-W fluorescent bulb (100-μs rise time, measured by a photocell connected to a storage oscilloscope) that was located 15 cm behind each stabilimeter. The unconditioned stimulus (US) was a 0.5-s, 0.4-mA shock produced by four LeHigh Valley shock generators (SGS-004; LeHigh Valley, Beltsville, MD) located outside the chamber and delivered to the floor bars of each stabilimeter. Shock intensity was measured with a 1-kΩ resistor across a differential channel of an oscilloscope in series with a 100-kΩ resistor connected between adjacent floor bars within each stabilimeter. Current was defined as the root mean square voltage across the 1-kΩ resistor, where mA equaled 0.707 × 0.5 × peak-to-peak voltage. The presentation and sequencing of all stimuli were under the control of the Macintosh G3 computer using specially designed software (The Experimenter, Glassbeads Inc., Newton, CT).
Behavioral Procedures and Drug Administration
Baseline startle test
On 2 consecutive days, animals were placed in the stabilimeter devices for 5 min without exposure to startle stimuli (acclimation period), followed by 15 startle-eliciting noise burts that were given in a semirandom order, 5 at each of three different intensities (95, 100, and 105 dB), with an interstimulus interval (ISI) of 30 s. These initial noise bursts were used to familiarize the rats to the acoustic stimuli and were not used for statistical analysis. Immediately after, 45 additional noise bursts were presented, 15 at each of the three aforementioned intensities with a 30-s ISI. The startle amplitudes across these 45 stimuli were used to divide rats into different test groups such that each group had equivalent mean startle amplitudes.
Fear-conditioning training
Twenty-four hours after the baseline startle test, rats were given two sessions of training, 24 h apart. In each of the training sessions, rats were placed in the stabilimeter devices and 5 min later animals were given 10 light-shock pairings that consisted of a 3.7-s light and a 0.5-s, 0.4-mA footshock that coterminates with the light. The mean intertrial interval (ITI) was 3 min (range: 2–4 min). Rats were returned to their home colony after completion of the training session.
Drug administration
Forty-eight hours after training, rats were bilaterally infused with either 0.2, 1.0, or 6.0 nmol (depending on the infusion site) of the SP receptor antagonist GR 82334 (Tocris Bioscience, MO) or artificial cerebrospinal fluid (ACSF—130 mM NaCl; 3.5 mM KCl; 1.10 mM KH2PO4; 1.3 mM MgCl2; 2.5 mM CaCl2; 30 mM NaHCO3; and 5.0 mM glucose). During the infusion, rats were placed in individual plastic cages (28 × 17 × 12 cm), the dummy stylets removed, and the infusion cannulas inserted (28 gauge, 1 mm projection from the tip of the guide cannula; Plastics One). The infusion cannulas were attached to Hamilton microsyringes (10 μl; Hamilton, Reno, NV) by polythylene tubing, and the drug solutions were delivered into the designated brain regions by a Harvard Apparatus pump (Model 22; Harvard Apparatus, South Natick, MA) at 0.25 μl/min for 2 min for a total 0.5 μl of drug solution in each infusion site. The infusion cannulas remained in place for an additional 2 min, after which they were removed and the dummy stylets replaced.
Fear-potentiated startle testing
Immediately after the infusion, rats were returned to the stabilimeters, where, after 5 min of acclimation, they were presented with 15 noise bursts identical to those in the baseline startle test. The response to these initial noise bursts was not used for statistical purposes. Following these initial noise bursts, 30 additional noise bursts (10 each at 95, 100, and 105 dB) were presented in the presence of the light (light–noise trials, LN) and 30 were presented in the absence of the light (noise-alone trials, NA) at an ISI of 30 s. On the light–noise trials, the startle stimulus occurred 3.2 s after the onset of the light, the same time point when the footshock had been delivered during training. The light–noise and noise-alone trials were presented in a balanced, pseudorandom order with the constraint that each trial type occurred only once in each consecutive six-trial block (light–noise or noise alone × 95, 100, and 105 dB).
Three days after the test session, rats with cannulations in the PAG or the dSC/DpMe were given one short retraining session consisting of five pairings of light and footshock with a 3-min ITI. During the ITIs, 10 noise bursts, 5 at each of 95 and 100 dB, were presented with a 60-s ISI. These noise bursts were used to minimize the possibility that the rats would learn to discriminate between training and test sessions based on the absence vs presence of noise bursts. Twenty-four hours later, the animals were infused with the AMPA receptor antagonist NBQX (disodium by Tocris, Avonmouth, Bristol, UK) or the opioid receptor agonist morphine sulfate (Sigma-Aldrich, St Louis, MO) and retested. We found that the retrained non-naive rats that were previously trained and, subsequently, infused with drug showed similar fear conditioning and drug response as did naive rats (Meloni and Davis, 1999; Zhao and Davis, 2004).
Histological Verification of Cannula Placement
At the end of the experiments, rats were deeply anesthesized by inhalation of Isoflurane, USP (Halocarbon, NJ) and were intracardially perfused with 0.9% saline followed by 8% buffered formalin. The brains were removed from the skull and immersed in a 30% sucrose–formalin solution for at least 3 days. Forty-micrometer coronal sections through the cannula track were prepared and every third section was mounted on gelatin-coated slides, air-dried, and stained with cresyl violet. The location of the cannulas was assessed under a light microscope and transcribed onto a brain atlas (Paxinos and Watson, 1997).
Statistical Analyses
Startle response amplitudes on either the noise-alone trials or the light–noise trials were averaged across the three noise burst intensities and served as the primary dependent variable (ie, one noise-alone value and one light–noise value for each rat). The difference in startle amplitude between the noise-alone and light–noise trials (between-trial difference) was also calculated as an additional dependent variable. Startle amplitude was analyzed using a two-way ANOVA (SPSS statistics package, Chicago, IL) with trial type (noise alone vs light–noise) and treatment group (dose) as within- and between-subject factors, respectively. Additionally one-way ANOVA followed by post hoc Dunnett's tests (two-sided) was used to compare the difference between the vehicle group and the drug-treated groups.
RESULTS
Cannula Placement
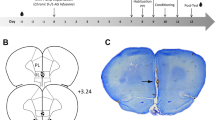
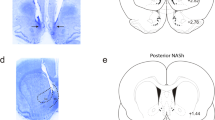
Figures 1 and 2 depict the cannula placements determined by the location of the tips of dummy cannulas. As shown in Figure 1, the cannulas were implanted into the MeA (the light gray circles), the CeA (the light gray squares), the BLA (the dark gray circles), and the VMH (the light open circles). Data from rats with cannulas that missed these areas were not included in the statistical analyses. Cannula implantations in the midbrain, as shown in Figure 2, included the rostral dlPAG (the dark gray circles), the caudal vlPAG (the black circles), and the dSC/DpMe (the dark gray squares). The location of the dSC/DpMe was described previously (Zhao and Davis, 2004), and includes the deep white and gray layers of the superior colliculus (DpWh and DpG, respectively, in Paxinos and Watson, 1997) and the dorsal part of the DpMe.

Cannula locations for different rats implanted with cannulas in the CeA; BLA; MeA; or the VMH (plates adapted from Paxinos and Watson, 1997).

Cannula locations for different rats implanted with cannulas in the dlPAG; the vlPAG; or the dSC/DpMe (plates adapted from Paxinos and Watson, 1997).
Role of Substance P in the Amygdala on the Expression of Fear-Potentiated Startle
In general, the ACSF-treated rats showed an increased startle response in the presence of the light cue that previously had been paired with a footshock. As shown in Figure 3, startle amplitude was higher on light–noise (open bar) than on noise-alone trials (closed bar) in the vehicle-treated animals. However, fear-potentiated startle was dose dependently reduced by the SP receptor antagonist, GR 82334, infused bilaterally into the MeA and the BLA, but not into the CeA. ANOVA revealed a significant main effect of trial type (noise alone vs light–noise) for the MeA (F(1, 31)=29.47, p<0.001), the BLA (F(1, 21)=23.43, p<0.001), and the CeA (F(1, 28)=24.75, p<0.001) groups. In addition, the trial type by treatment interaction was also significant for the MeA (F(2, 31)=5.49, p<0.01) and BLA groups (F(2, 21)=6.77, p<0.01), but not for the CeA group (F(2, 28)=1.02, p>0.05). Moreover, Dunnett's tests on the difference scores (ie, LN minus NA startle amplitude) indicated significant differences between the vehicle and high-dose GR 82334 groups that received infusions into the MeA (p<0.01) or BLA (p<0.05), but not into the CeA (p>0.05).

Mean amplitude of the startle reflex in the absence (black bars) or the presence (white bars) of the visual CS, or the difference between these two trial types (gray bars=fear-potentiated startle) after local infusion of various doses of the SP antagonist GR 82334 into either the MeA, the BLA, or the CeA.
Role of Substance P in the Medial Hypothalamus on the Expression of Fear-Potentiated Startle
Infusion of 1.0 nmol of GR 82334 into the medial hypothalamus did not disrupt fear-potentiated startle (Figure 4). When the dose was increased to 6.0 nmol, the expression of fear-potentiated startle was blocked with infusion into the VMH. However, when infused into the DMH, it was immediately noticed that this caused remarkable locomotor changes that included severe body flipping, rotation, and bending of the head forward. These deficits were not seen with infusion into the VMH. We have frequently observed that profound locomotor changes caused by drug infusion into certain brain areas interfere with the expression of fear-potentiated startle. Thus, GR 82334 infusions into the DMH were terminated. With respect to the VMH, ANOVA revealed a significant main effect of trial type (F(1, 28)=24.75, p<0.001) and a significant trial type by treatment interaction (F(2, 28)=3.36, p<0.05), and Dunnett's tests indicated significant differences between the vehicle group and the 6-nmol group (p<0.01).

Mean amplitude of the startle reflex in the absence (black bars) or the presence (white bars) of the visual CS, or the difference between these two trial types (gray bars=fear-potentiated startle) after local infusion of various doses of the SP antagonist GR 82334 into either the VMH or DMH.
Role of Substance P in the Midbrain on the Expression of Fear-Potentiated Startle
Infusion of 1 nmol GR 82334 into the rostral dlPAG (Figure 5, upper panel) abolished fear-potentiated startle. Statistically, this was indicated by an ANOVA that revealed a significant trial type by treatment interaction, F(1, 12)=6.87, p<0.05. Anatomical specificity was indicated by the ineffectiveness of infusions into two nearby areas. Specifically, infusion of the same dose into the caudal vlPAG (Figure 5, middle panel) had no effect on fear-potentiated startle (trial type by treatment interaction—F(1, 16)=0.72, p>0.05), nor did infusion into the dSC/DpMe (trial type by treatment interaction—F(1, 10)=0.92, p>0.05), which is approximately 1 mm lateral to the rostral dlPAG (Figure 5, lower panel).

Mean amplitude of the startle reflex in the absence (black bars) or the presence (white bars) of the visual CS, or the difference between these two trial types (gray bars=fear-potentiated startle) after local infusion of various doses of the SP antagonist GR 82334 into either rostral part of the dlPAG; the caudal part of the vlPAG; or the dSC/DpMe.
Our laboratory previously found that infusion of the AMPA receptor antagonist NBQX into the dSC/DpMe, but not the rostral dlPAG, blocks fear-potentiated startle (Zhao and Davis, 2004). Recently, we also found that infusion of the opioid receptor agonist morphine into the caudal vlPAG similarly blocks potentiated startle (Davis et al, in preparation). Therefore, as a positive control for the negative effect of GR 82334 in these two areas, these rats were retrained, and retested with infusions of NBQX into the dSC/DpMe or into the rostral dlPAG, or with infusion of morphine into the caudal vlPAG. The results are shown in Figure 6. As depicted in the upper and the middle panels, infusion of NBQX (0.79 nmol) into the dSC/DpMe, but not the rostral PAG blocked fear-potentiated startle (trial type by treatment interactions—for the dSC/DpMe infusions, and F(1, 12)=0.052, p>0.05 for the rostral dlPAG infusions, respectively) and morphine infusions (20 nmol) into the caudal vlPAG were similarly effective (trial type by treatment interaction—F(1, 14)=5.86, p<0.05, lower panel).

Mean amplitude of the startle reflex in the absence (black bars) or the presence (white bars) of the visual CS, or the difference between these two trial types (gray bars=fear-potentiated startle) after local infusion of the AMPA receptor antagonist into the rostral part of the dlPAG, the dSC/DpMe, or after morphine in the caudal part of the vlPAG.
DISCUSSION
Blockade of SP receptors in the MeA and the BLA severely disrupted fear-potentiated startle, whereas similar doses in the CeA had no detectable effect, consistent with the existing literature. Ebner et al (2004) showed that immobilization stress caused a significant increase in SP release within the MeA but not the CeA. They also found that intra-MeA SP infusions produced anxiety-like behaviors as assessed with elevated plus maze, an effect similar to that induced by immobilization. In the BLA, immobilization or maternal separation produced an increase in SP receptor internalization, a marker of SP release (Boyce et al, 2001; Smith et al, 1999), and intra-BLA infusion of an SP receptor antagonist significantly attenuated separation-induced vocalizations (Boyce et al, 2001).
It is widely believed that conditioned fear stimuli activate BLA projection neurons which send afferents to various downstream target areas such as the CeA and MeA (Davis, 1992) (LeDoux, 1992). Activation of SP receptors leads to postsynaptic excitation via G-protein-coupled SP receptors, which stimulate phospholipase C and adenylate cyclase (Mitsuhashi et al, 1992; Otsuka and Yoshioka, 1993; Takeda et al, 1992). Our finding that fear-potentiated startle could be blocked by infusing an SP receptor antagonist into the BLA suggests that SP receptor activation contributes to or is required for activation of BLA projection neurons. Interestingly, however, it has been found that SP receptors in the BLA are largely, if not exclusively, expressed in interneurons (Levita et al, 2003; Maubach et al, 2001). Application of SP onto BLA slices induces excitation of these interneurons and this, in turn, hyperpolarizes adjacent projection neurons via GABAA receptors (Maubach et al, 2001). By interfering with the normal balance between excitation and inhibition, an SP receptor antagonist may sufficiently alter network properties so as to disrupt the normal response to fear-signaling stimuli.
One target of BLA output neurons is the MeA. Although there are reciprocal connections between the BLA and MeA (Pitkanen et al, 1997), the fact that sensory inputs converge in the BLA but not in the MeA (Jolkkonen et al, 2001; LeDoux, 2000; Shi and Davis, 2001) suggests that, for fear expression, the MeA may function primarily as an output structure that responds to fear-related signals arriving from the BLA. Our previous findings that pretraining infusions of NBQX into the BLA, but not into the MeA, blocked fear learning, whereas pretest infusions into both structures blocked fear expression are consistent with this view (Walker et al, 2005). Given that BLA projection neurons are largely glutamatergic (Smith and Pare, 1994) and that there are few SP-containing neurons in the BLA (Ribeiro-da-Silva and Hokfelt, 2000; Roberts et al, 1982), it is reasonable to assume that neurotransmission between the BLA and the MeA is primarily glutamatergic, and not tachykininergic. A double labeling study using a retrograde tracer injected into the MeA combined with immunostaining for SP would be an important initial step in identifying the site of origin of SP inputs to the MeA that participates in fear expression. The MeA itself contains a large number of neurons that contain SP, and it is possible that NK1 receptors in the MeA are responding, at least in part, to locally released SP. Speculatively, SP neurons within the MeA may be activated by glutamatergic projections from the BLA.
The CeA is also a major recipient of BLA projections and is believed to play an important role in fear-potentiated startle, based partly on evidence that CNQX or NBQX infusions into the CeA block fear-potentiated startle (Kim et al, 1993; Walker and Davis, 1997). However, in the present study, doses of GR 82334 that blocked fear-potentiated startle when infused into the BLA or the MeA did not significantly disrupt fear-potentiated startle when infused into the CeA. Insofar as the CeA is situated immediately medial to the BLA and just lateral and dorsal to the MeA, this negative finding serves as an important control in establishing the fact that the effect of GR 82334 infusions into the BLA or MeA were mediated locally and not by diffusion to nearby areas. Of course, it is possible that higher doses might have affected fear-potentiated startle. However, pilot data from several animals, which received intra-CeA infusions of 5 nmol of GR 82334, revealed marked locomotor effects that precluded an analysis of fear-potentiated startle.
Early studies have shown that the VMH also is involved in fear and anxiety (Colpaert and Wiepkema, 1976; de Oliveira et al, 1997; Dielenberg et al, 2001; Han et al, 1996; Sudakov, 1987) and, interestingly, the VMH itself receives a heavy SP-containing input from the MeA (Petrovich et al, 2001; Shaikh et al, 1993). In fact, this pathway has previously been shown to be critical in mediating defensive rage and predatory attack in cats (Han et al, 1996; Shaikh et al, 1993). We found that GR 82334 infusions into the VMH also blocked fear-potentiated startle. It is possible that SP-containing neurons in the MeA use the VMH as a relay through which the influences of the MeA on fear-potentiated startle are mediated. It should be noted, however, that a higher dose of GR 82334 was required in the VMH compared with the BLA or the MeA. This may be due, in part, to the differential expression of SP receptor subtypes in the amygdala compared with hypothalamus. Thus, both septide and non-septide-sensitive SP receptors are expressed in the amygdala and activation of either induces intracellular signaling. However, in the hypothalamus, the primary SP receptors are the non-septide-sensitive SP receptors to which GR 82334 has a lower affinity (Beaujouan et al, 2000). Therefore, a higher dose of GR 82334 may be required to block SP neurotransmisson in the VMH.
A role for the PAG in mediating fear and anxiety has also been well documented (Bandler and Shipley, 1994; Blanchard et al, 1981; DeOca et al, 1998) and SP neurotransmission has been implicated here as well (De Araujo et al, 1999; Gregg and Siegel, 2003; Mongeau et al, 1998). The findings of our study support a critical role of SP neurotransmission within the PAG in fear-potentiated startle and indicate more specifically that these effects may be mediated primarily by SP receptors in the rostral dlPAG. Perhaps not by coincidence, this is a region that receives a dense projection from the VMH (Bandler and Shipley, 1994; Canteras et al, 1994; Siegel et al, 1997), and approximately 30% of these projections contain SP (Dornan et al, 1990). However, the projections outlined in this paper went to a significantly more caudal region of the dlPAG than was targeted by any of the injections performed as part of the present study. Thus, it is possible that SP release from VMH terminals in the PAG is critically involved in the expression of fear-potentiated startle. However, it is also possible that local SP circuit neurons in the PAG are involved instead or as well. The VMH also projects to the vlPAG, particularly, to its more caudal elements (Canteras et al, 1994), but it is unknown whether these projections also contain SP. Although SP receptors are distributed throughout the PAG (Barbaresi, 1998; Commons and Valentino, 2002), our data indicate that those in the caudal vlPAG play a less significant role in fear-potentiated startle.
Presently, it is not clear why SP antagonists infused into the BLA, MeA, VMH, and PAG so completely block fear-potentiated startle. There are at least two major questions in this regard. First, it is not clear how inactivation of apparently parallel neural pathways (eg, BLA → CeA → dSC/DpMe and BLA → MeA → VMH → PAG) each completely block the expression of fear-potentiated startle. Instead, one might expect that either pathway could mediate fear-potentiated startle alone, so that inactivation of points along both pathways, rather than just one, would be required to totally block fear-potentiated startle. Second, it is not clear how SP might be acting in the BLA, MeA, VMH, and PAG to affect fear-potentiated startle. Although local infusion of SP into each of these sites can produce defensive and anxiety-like behaviors, we did not see any increase in startle when we infused SP into the MeA over a very wide range of doses (Davis et al, in preparation). Indeed, at high doses, SP actually interfered with fear-potentiated startle, perhaps via internalization of the SP receptor. However, the inability of SP to facilitate startle does not necessarily rule out the idea that release of SP is involved in mediating fear-potentiated startle, because we clearly cannot recreate the temporal dynamics of endogeous SP release by flooding the system with exogenous SP.
In a long series of pilot studies (Davis et al, in preparation), we were not able to demonstrate convincingly a serial circuit from the MeA to VMH vis-a-vis SP. For example, although local infusion of GR 82334 into the left MeA or right VMH (or vice versa) by themselves did not fully block fear-potentiated startle, infusion into the left MeA and right VMH did, consistent with a serial circuit. However, equivalent doses of GR 82334 into the left MeA and the left VMH together also did, inconsistent with a serial process. It is, thus, possible that SP plays a more permissive role in these structures. For example, in the spinal cord, SP has no effect by itself, but is a powerful modulator of both NMDA- and AMPA-mediated excitation of nociceptive neurons in the spinal cord (Budai et al, 1992).
Taken together with findings from our laboratory and elsewhere, we propose that fear-potentiated startle is mediated by the amygdala through two separate neural pathways (see Figure 7). The first one originates in the CeA and connects to the PnC via a synapse in the dSC/DpMe of the rostral midbrain. This pathway appears to rely primarily on ionotropic glutamatergic transmission at the dSC/DpMe. The second pathway, as suggested by the findings reported here, originates in the MeA and may access the primary startle reflex circuit using relays located in the VMH and rostral dlPAG. This pathway appears to require NK1 receptor activation. Interestingly, blockade of glutamate receptors (Zhao and Davis, 2004) or activation of GABA-A receptors (Meloni and Davis, 1999) in the dSC/DpMe, or blockade of SP receptors or activation of opioid receptors (Davis et al, in preparation) in the MeA, VMH, and PAG each totally blocks the expression of fear-potentiated startle. Thus, although these pathways represent parallel outputs connecting the amygdala with the PnC, collectively, the data suggest that activation of each is necessary for the expression of fear-potentiated startle under normal circumstances. In addition, anatomically, the CeA projects directly to the PnC based on anterograde and retrograde tracing (Rosen et al, 1991), indicating that a third pathway might also be involved in the expression of fear-potentiated startle. However, to date, there is no direct evidence that this pathway is actually involved because its role was tested using electrolytic lesions at various levels along this pathway (Hitchcock and Davis, 1991), which could have interfered with the indirect pathways via the dSC/DpMe or the VMH and PAG.

A schematic diagram that illustrates the hypothesized neural pathways through which the amygdala may mediate fear-potentiated startle. Fear is induced by the presence of the conditioned stimulus (CS), which was previously paired with the unconditioned stimulus (US), and measured by an increase in startle amplitude elicited by a loud noise in the presence of the CS. The fear-potentiated startle is mediated by the amygdala through two separate pathways, namely the central amygdalo-tegmento-pontine pathway and the medial amygdalo-hypothalamo-periaqueducto-pontine pathway. The central amygdalo-tegmento-pontine pathway involves the central nucleus of the amygdala (CeA), the deep layers of the superior colliculus/deep mesencephalic nucleus (dSC/DpMe) in the midbrain, and the nucleus reticularis pontis caudalis (PnC) of the brainstem. Glutamatergic neurotransmission is required in this pathway. The medial amygdalo-hypothalamo-periaqueducto-pontine pathway involves the medial nucleus of the amygdala (MeA), the ventromedial nucleus of the hypothalamus (VMH), the dorsolateral periaqueductal gray (dlPAG) in the midbrain and the PnC. In this circuit, substance P neurotransmission is required, although we do not know how. We hypothesize that it modulates the release of glutamate in some but not all of these nuclei, including the PnC, based on the work of Kungel et al (1994). The startle circuit consists of cochlear root neurons (CN) in the auditory nerve, the PnC, and motor neurons in the facial motor nucleus and spinal cord, where a startle stimulus activates CNs and subsequently elicits a rapid startle reflex. The CeA also projects directly to the PnC which may, via temporal summation with other inputs, increase the excitability of the PnC.
Finally, SP also plays an important role at the level of the PnC in the startle pathway. Kungel et al (1994) found that acoustically responsive neurons in the PnC showed a marked increase in firing rate following iontophoretic injection of SP and local infusion of SP into the PnC caused a dose-dependent increase in startle amplitude (Krase et al, 1994). This increase in startle, as well as shock sensitization of startle, was blocked by local infusion of an SP antagonist here. In this case, the source of SP appears to come from the laterodorsal tegmental nucleus and the pedunculopontine tegmental nucleus (Kungel et al, 1994).
References
Bandler R, Shipley MT (1994). Columnar organization in the midbrain periaqueductal gray: modules for emotional expression? Trends Neurosci 17: 379–389.
Barbaresi P (1998). Immunocytochemical localization of substance P receptor in rat periaqueductal gray matter: a light and electron microscopic study. J Comp Neurol 398: 473–490.
Beaujouan JC, Saffroy M, Torrens Y, Glowinski J (2000). Different subtypes of tachykinin NK(1) receptor binding sites are present in the rat brain. J Neurochem 75: 1015–1026.
Blanchard DC, Williams G, Lee EMC, Blanchard RJ (1981). Taming of wild Rattus norvegicus by lesions of the mesencephalic central gray. Physiol Behav 9: 157–163.
Boyce S, Smith D, Carlson E, Hewson L, Rigby M, O'Donnell R et al (2001). Intra-amygdala injection of the substance P (NK(1) receptor) antagonist L-760735 inhibits neonatal vocalisations in guinea-pigs. Neuropharmacology 41: 130–137.
Budai D, Wilcox GL, Larson AA (1992). Modulation of N-methyl-D-aspartate and (R,S)-alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) responses of spinal nociceptive neurons by a N-terminal fragment of substance P. Eur J Pharmacol 216: 441–444.
Cameron AA, Iqbal AK, Westlund KN, Willis WD (1995). The efferent projections of the periaqueductal gray in the rat: a Phaseolus vulgaris-leucoagglutinin study. II. Descending projections. J Comp Neurol 351: 585–601.
Canteras NS, Simerly RB, Swanson LW (1994). Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol 348: 41–79.
Canteras NS, Simerly RB, Swanson LW (1995). Organization of projections from the medial nucleus of the amygdala: a PHAL study in the rat [published erratum appears in J Comp Neurol 1996; 369(2):328–330]. J Comp Neurol 360: 213–245.
Cassella JV, Davis M (1986). The design and calibration of a startle measurement system. Physiol Behav 36: 377–383.
Castellano C, McGaugh JL (1989). Retention enhancement with post-training picrotoxin: lack of state dependency. Behav Neural Biol 51: 165–170.
Colpaert FC, Wiepkema PR (1976). Effects of ventromedial hypothalamic lesions on spontaneous intraspecies aggression in male rats. Behav Biol 16: 117–125.
Commons KG, Valentino RJ (2002). Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J Comp Neurol 447: 82–97.
Damalama M, Swann J (1993). Substance P and neurokinin A are colocalized in the central chemosensory pathway of the male golden hamster. Neuropeptides 24: 327–334.
Davis M (1992). The role of the amygdala in fear and anxiety. Ann Rev Neurosci 15: 353–375.
Davis M, Falls WA, Campeau S, Kim M (1993). Fear-potentiated startle: a neural and pharmacological analysis. Behav Brain Res 58: 175–198.
Davis M, Walker DL, Lee Y (1997). Roles of the amygdala and bed nucleus of the stria terminalis in fear and anxiety measured with the acoustic startle reflex: possible relevance to PTSD. Ann NY Acad Sci 821: 305–331.
Davis M, Shi C-J, Yang Y, Zhao Z . Effects of morphine in the amygdala, ventromedial hypothalamus and periaqueductal grey on fear potentiated startle (in preparation).
De Araujo JE, Silva RC, Huston JP, Brandao ML (1999). Anxiogenic effects of substance P and its 7–11 C terminal, but not the 1–7 N terminal, injected into the dorsal periaqueductal gray. Peptides 20: 1437–1443.
de Oliveira L, Hoffmann A, Menescal-de-Oliveira L (1997). The lateral hypothalamus in the modulation of tonic immobility in guinea pigs. NeuroReport 8: 3489–3493.
DeOca BM, DeCola JP, Maren S, Fanselow MS (1998). Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci 18: 3426–3432.
Dielenberg RA, Hunt GE, McGregor IS (2001). ‘When a rat smells a cat’: the distribution of Fos immunoreactivity in rat brain following exposure to a predatory odor. Neurosci 104: 1085–1097.
Dornan WA, Akesson TR, Micevych PE (1990). A substance P projection from the VMH to the dorsal midbrain central gray: implication for lordosis. Brain Res Bull 25: 791–796.
Ebner K, Rupniak NM, Saria A, Singewald N (2004). Substance P in the medial amygdala: emotional stress-sensitive release and modulation of anxiety-related behavior in rats. Proc Natl Acad Sci USA 101: 4280–4285.
Emson PC, Jessell T, Paxinos G, Cuello AC (1978). Substance P in the amygdaloid complex, bed nucleus and stria terminalis of the rat brain. Brain Res 149: 97–105.
Fendt M, Koch M, Schnitzler H-U (1994). Lesions of the central gray block the sensitization of the acoustic startle response in rats. Brain Res 661: 163–173.
Frankland PW, Yeomans JS (1995). Fear-potentiated startle and electrically evoked startle mediated by synapses in rostrolateral midbrain. Behav Neurosci 109: 669–680.
Gregg TR, Siegel A (2003). Differential effects of NK1 receptors in the midbrain periaqueductal gray upon defensive rage and predatory attack in the cat. Brain Res 994: 55–66.
Han Y, Shaikh MB, Siegel A (1996). Medical amygdaloid suppression of predatory attack behavior in the cat: I. Role of a substance P pathway from the medial amygdala to the medial hypothalamus. Brain Res 716: 59–71.
Hitchcock JM, Davis M (1991). The efferent pathway of the amygdala involved in conditioned fear as measured with the fear-potentiated startle paradigm. Behav Neurosci 105: 826–842.
Jolkkonen E, Pikkarainen M, Kemppainen S, Pitkanen A (2001). Interconnectivity between the amygdaloid complex and the amygdalostriatal transition area: a PHA-L study in rat. J Comp Neurol 431: 39–58.
Kim M, Campeau S, Falls WA, Davis M (1993). Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 59: 5–8.
Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ et al (1998). Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 281: 1640–1645.
Krase W, Koch M, Schnitzler HU (1994). Substance P is involved in the sensitization of the acoustic startle response by footshock in rats. Behav Brain Res 63: 81–88.
Kungel M, Ebert U, Herbert H, Ostwald J (1994). Substance P and other putative transmitters modulate the activity of reticular pontine neurons: an electrophysiological and immunohistochemical study. Brain Res 643: 29–39.
LeDoux JE (1992). Emotion and the amygdala. In: Aggleton JP (ed). The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. Wiley-Liss: New York. pp 339–352.
LeDoux JE (2000). The amygdala and emotion: a view through fear. In: Aggleton JP (ed). The Amygdala. Oxford University Press: New York. pp 289–310.
Levita L, Mania I, Rainnie DG (2003). Subtypes of substance P receptor immunoreactive interneurons in the rat basolateral amygdala. Brain Res 981: 41–51.
Malsbury CW, McKay K (1989). Sex difference in the substance P-immunoreactive innervation of the medial nucleus of the amygdala. Brain Res Bull 23: 561–567.
Mantyh PW (2002). Neurobiology of substance P and the NK1 receptor. J Clin Psychiat 63 (Suppl 11): 6–10.
Maubach KA, Martin K, Smith DW, Hewson L, Frankshun RA, Harrison T et al (2001). Substance P stimulates inhibitory synaptic transmission in the guinea pig basolateral amygdala in vitro. Neuropharmacology 40: 806–817.
Meloni EG, Davis M (1999). Muscimol in the deep layers of the superior colliculus/mesencephalic reticular formation blocks expression but not acquisition of fear-potentiated startle in rats. Behav Neurosci 113: 1152–1160.
Mitsuhashi M, Osashi Y, Shichijo S, Christian C, Sudduth-Klinger J, Harrowe G et al (1992). Multiple intracellular signaling pathways of the neuropeptide substance P receptor. J Neurosci Res 32: 437–443.
Mongeau R, De Oca BM, Fanselow MS, Marsden CA (1998). Differential effects of neurokinin-1 receptor activation in subregions of the periaqueductal gray matter on conditional and unconditional fear behaviors in rats. Behav Neurosci 112: 1125–1135.
Otsuka M, Yoshioka K (1993). Neurotransmitter functions of mammalian tachykinins. Physiological Rev 73: 229–308.
Paxinos G, Watson C (1997). The Rat Brain in Stereotaxic Coordinates, 3rd edn. Academic Press: New York.
Petrovich GD, Canteras NS, Swanson LW (2001). Combinatorial amygdalar inputs to hippocampal domains and hypothalamic behavior systems. Brain Res Brain Res Rev 38: 247–289.
Pitkanen A, Savander V, LeDoux JE (1997). Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20: 517–523.
Ribeiro-da-Silva A, Hokfelt T (2000). Neuroanatomical localisation of Substance P in the CNS and sensory neurons. Neuropeptides 34: 256–271.
Roberts GW, Woodhams PL, Polak JM, Crow TJ (1982). Distribution of neuropeptides in the limbic system of the rat: the amygdaloid complex. Neurosci 7: 99–131.
Rosen JB, Hitchcock JM, Sananes CB, Miserendino MJD, Davis M (1991). A direct projection from the central nucleus of the amygdala to the acoustic startle pathway: anterograde and retrograde tracing studies. Behav Neurosci 105: 817–825.
Rosenkranz MA (2007). Substance P at the nexus of mind and body in chronic inflammation and affective disorders. Psychol Bull 33: 1007–1037.
Saffroy M, Beaujouan JC, Torrens Y, Besseyre J, Bergstrom L, Glowinski J (1988). Localization of tachykinin binding sites (NK1, NK2, NK3 ligands) in the rat brain. Peptides 9: 227–241.
Sergeyev V, Fetissov S, Mathe AA, Jimenez PA, Bartfai T, Mortas P et al (2005). Neuropeptide expression in rats exposed to chronic mild stresses. Psychopharmacol 178: 115–124.
Shaikh MB, Steinberg A, Siegel A (1993). Evidence that substance P is utilized in medial amygdaloid faciliation of defensive rage behavior in the cat. Brain Res 625: 283–294.
Shi C-J, Davis M (2001). Visual pathways involved in fear conditioning measured with fear-potentiated startle: behavior and anatomic studies. J Neurosci 21: 9844–9855.
Siegel A, Schubert KL, Shaika MB (1997). Neurotransmitters regulating defensive rage behavior in the cat. Neurosci Biobehav Rev 21: 733–742.
Smith DW, Hewson L, Fuller P, Williams AR, Wheeldon A, Rupniak NM (1999). The substance P antagonist L-760,735 inhibits stress-induced NK(1) receptor internalisation in the basolateral amygdala. Brain Res 848: 90–95.
Smith Y, Pare D (1994). Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with postembedding GABA and glutamate immunocytochemistry. J Comp Neurol 342: 232–248.
Sudakov KV (1987). Angiotensin II as a factor inhibiting the fear response. Neurosci Behav Physiol 17: 74–83.
Takeda Y, Blount P, Sachais BS, Hershey AD, Raddatz R, Krause JE (1992). Ligand binding kinetics of substance P and neurokinin A receptors stably expressed in Chinese hamster ovary cells and evidence for differential stimulation of inositol 1,4,5-trisphosphate and cyclic AMP second messenger responses. J Neurochem 59: 740–745.
Walker DL, Davis M (1997). Double dissociation between the involvement of the bed nucleus of the stria terminalis and the central nucleus of the amygdala in light-enhanced versus fear-potentiated startle. J Neurosci 17: 9375–9383.
Walker DL, Paschall GY, Davis M (2005). Glutamate receptor antagonist infusions into the basolateral and medial amygdala reveal differential contributions to olfactory vs. context fear conditioning and expression. Learn Mem 12: 120–129.
Zhao Z, Davis M (2004). Fear-potentiated startle in rats is mediated by neurons in the deep layers of the superior colliculus/deep mesencephalic nucleus of the rostral midbrain through the glutamate non-NMDA receptors. J Neurosci 24: 10326–10334.
Acknowledgements
This research was supported by National Institute of Mental Health Grants MH47840, MH57250, and MH59906; the Science and Technology Center (the Center for Behavioral Neuroscience of the National Science Foundation under Agreement no. IBN-9876754); and the Yerkes Base Grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
The authors have no conflict of interest to report related to the finding in this study. However, Dr Michael Davis does have a project on a grant from the NIMH (U19 MH06905602) to test compounds supplied by GlaxoSmithKline, one of which is an NK1 antagonist.
Michael Davis
SAB Helicon Corp.—now terminated
Consultant, Repligen Corp.—2002–2005
Consultant, Amgen Corp.—2 January 2004
Consultant, AstraZeneca—6 June 2006
Consultant, Tikvah Therapeutics Inc.—2007–present
Research support—Repligen Corp.—2002–2005
Research support—AstraZeneca
Unrestricted gift—AstraZeneca
Dr Davis has a patent pending for the use of cognitive enhancers, in particular, D-cycloserine, to be used as an adjunct to psychotherapy. Tikvah Therapeutics Inc. has licensed this technology to commercialize the use of D-cycloserine as an adjunct to psychotherapy. He is a partner in a company called Therapade and has received a signing fee. He will then receive certain milestone payments over the next several years. Once the indication is approved by the FDA, he is entitled to royalties.
Rights and permissions
About this article
Cite this article
Zhao, Z., Yang, Y., Walker, D. et al. Effects of Substance P in the Amygdala, Ventromedial Hypothalamus, and Periaqueductal Gray on Fear-Potentiated Startle. Neuropsychopharmacol 34, 331–340 (2009). https://doi.org/10.1038/npp.2008.55
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2008.55
Keywords
This article is cited by
-
A Deep Mesencephalic Nucleus Circuit Regulates Licking Behavior
Neuroscience Bulletin (2022)
-
The associations of TAC1 gene polymorphisms with major depressive disorder
Molecular & Cellular Toxicology (2019)
-
Overlapping expression of serotonin transporters and neurokinin-1 receptors in posttraumatic stress disorder: a multi-tracer PET study
Molecular Psychiatry (2016)
-
Increased neurokinin-1 receptor availability in the amygdala in social anxiety disorder: a positron emission tomography study with [11C]GR205171
Translational Psychiatry (2015)
-
The brain orexin system and almorexant in fear-conditioned startle reactions in the rat
Psychopharmacology (2012)
