
Abstract
The much sought after optical transistor—the photonic counterpart of the electronic transistor—is poised to become a central ingredient in the development of optical signal processing. The motivation for using photons rather than electrons comes not only from their faster dynamics, but also from their lower crosstalk and robustness against environmental decoherence, which enable a high degree of integration and the realization of quantum operations1. A single-molecule transistor has recently been demonstrated at cryogenic temperatures2. Here, we demonstrate that a single nitrogen–vacancy centre at room temperature can operate as an optical switch under non-resonant continuous-wave illumination. We show an optical modulation of more than 80% and a time response faster than 100 ns in the green-laser-driven fluorescence signal, which we control through an independent near-infrared gating laser. Our study indicates that the near-infrared laser triggers a fast-decay channel of the nitrogen–vacancy mediated by promotion of the excited state to a dark band.
Similar content being viewed by others

Main
Unlike charged particles such as electrons, photons interact extremely weakly with each other3. Therefore, an optical switch requires the mediation of a physical system to produce efficient photon–photon interactions. Different approaches to optical transistors have been proposed4,5,6, often based on nonlinearities in well-defined photonic resonators. More recently, a high-finesse optomechanical resonator has been used to demonstrate a transistor-like effect7. The ultimate goal for future optical commutation technologies is to rely on single atoms or molecules having the ability to manipulate light down to the single-photon level, which feature an intrinsically high nonlinearity and grant us access to exploiting the quantum nature of light8. In this direction, single-molecule optical processing has recently been achieved at low temperatures2. Furthermore, the inclusion of a third, dark state in the electronic structure of the atom or molecule has been argued to be particularly advantageous to reduce the effect of thermal decoherence9,10. The quest for operating such optical nano-transistors at room temperature is attracting considerable attention, as it would have a major impact on photonic technologies and enable high-speed signal processing in general.
In parallel, research on single quantum emitters (either molecules or quantum dots) has focused on achieving stable and efficient light emission with well-defined properties11,12,13,14. However, a long-term stable source of single photons at room temperature still remains challenging with these emitters. In this context, nitrogen–vacancy centres in diamond have recently been intensely investigated owing to their emission stability. Nitrogen–vacancy centres are artificial atoms protected from their environment by a diamond shell, and consequently, they are immune to both blinking and bleaching, so that their electron spin features a long coherence time, even at room temperature15. Their suitability for quantum-optics experiments has opened a new route towards the implementation of solid-state quantum simulators at room temperature16,17. Despite the sustained effort to study the properties of nitrogen–vacancy centres over the past decade18, new insights into their energy states are still emerging19,20. Here, we describe an unconventional configuration in which a single nitrogen–vacancy operates as an all-optical nano-modulator at room temperature on non-resonant continuous-wave near-infrared (NIR) illumination. Besides measuring and characterizing the switch effect, we provide insight into the mechanism responsible for the nitrogen–vacancy nonlinearity by revealing the presence of a dark band that can be optically activated with an infrared laser. This new process is successfully described by a rate-equation model, further allowing us to predictively simulate and optimize the effect, which could be interesting for optical quantum technologies based on nitrogen–vacancy centres.
Our concept of an optical modulator based on a single nitrogen–vacancy centre is sketched in Fig. 1. The nitrogen–vacancy centre has a steady fluorescence (output signal) when pumped with a continuous-wave green laser (input signal). Illumination with a non-resonant continuous-wave NIR laser (gate signal) drives the excited state back to the ground state through non-radiative decay. In the following, we experimentally demonstrate the viability of this new gating channel, and speculate the potential involvement of a dark band of states to which the NIR laser promotes the excited state. This mechanism thus opens the possibility of controlling the number of emitted fluorescence photons by adjusting the intensity of the NIR gating laser.

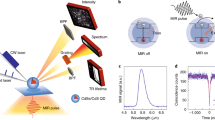
a,b, As for its electronic counterpart (a), the emitter signal of the optical switch (nitrogen–vacancy fluorescence on continuous green-laser illumination) is controlled by a gate provided by a modulated non-resonant NIR laser (b). c, In absence of the NIR laser, the nitrogen-vacancy can be regarded as a two level system excited by a green pumping beam. d, The NIR laser populates a dark band D, which decays non-radiatively, thus reducing the fluorescence output (d) compared with the emission without NIR laser (c).
In our experiment, 70 nm nanodiamonds are dispersed on a glass coverslip that is placed in the sample plane of a scanning confocal fluorescence microscope. The nitrogen–vacancy centres are pumped with a 532 nm continuous-wave laser (input beam) after focusing through an immersion objective lens (NA = 1.2), and their fluorescence (output beam) is collected back through the same objective and sent to an avalanche photodiode (Fig. 2a). Hanbury-Brown and Twist measurements enable us to identify a nanodiamond that hosts a single nitrogen–vacancy (see Supplementary Information). The 1,064 nm laser beam (gating beam) is superimposed with the 532 nm beam after passing through an acousto-optic modulator (AOM) that allows us to temporally modulate its power (see Fig. 2c,d).

a, A green laser (λ = 532 nm) is focused through a high-numerical-aperture objective (NA = 1.2) to excite a single nitrogen–vacancy (NV) centre in a nanodiamond crystal deposited on a glass coverslip (refractive index nglass = 1.52). A NIR laser (λ = 1,064 nm, shown in red in the sketch) is super-imposed to the 532 nm laser. The emitted fluorescence (orange) passes through a dichroic filter and is detected with two avalanche photodiodes. b, Setting the excitation power of the green laser to 55 μW while increasing the power of the NIR laser, a 1/(1+PNIR/Psat) fluorescence decrease is observed, which is linear for small power (inset) and nonlinear when PNIR increases. c,d, By modulating the gating NIR laser with an AOM for constant green-laser excitation power, a mimicking modulation of the nitrogen–vacancy fluorescence is observed. Tuning the power of the gating laser pulses allows us to reach different fluorescence decay levels (c). Here, we demonstrate the possibility of defining at least 8 states (that is, well-differentiated output levels) for potential application as a digital optical switch. The measured emission from the nitrogen–vacancy for different gating laser pulse lengths ranging from 3 μs down to 200 ns illustrates the time response of the system (d). In our configuration, the response is faster than 200 ns and is limited by the electronic time response of our AOM.
We start by examining the effect of 1,064 nm illumination on the fluorescence yield of a single nitrogen–vacancy centre under constant pumping with a 55 μW green light. The NIR power dependence of the fluorescence counts plotted in Fig. 2b shows a monotonous drop that is apparently linear below 15 mW (inset of Fig. 2b) before reaching saturation. This demonstrates the possibility of modulating the fluorescence emission from a single nitrogen–vacancy by up to 80% for this chosen green excitation power. Quenching of fluorescence could be obtained with other molecular species by means of alternative mechanisms such as conformational transformation in photochromic molecules21,22, although these are intrinsically much slower than the electronic response time of our system. Nevertheless, unlike molecules or quantum dots11,13, nitrogen–vacancy centres are stable and bright emitters, allowing us to observe this effect at room temperature and over a long time span15,18. A decrease in the fluorescence of nitrogen–vacancy centres has been observed in previous studies on stimulated emission depletion under resonant excitation23, multiphoton absorption induced by high-power pulsed lasers24, local heating to hundreds of degrees25, or even change of the nitrogen–vacancy charge state26). In contrast, we here invoke a radically different physical mechanism, as emphasized by the fact that our non-resonant continuous-wave NIR illumination of a few tens of milliwatts cannot trigger any of the above-mentioned mechanisms. Figure 2c shows the evolution of fluorescence from the same nitrogen–vacancy centre while applying a sequence of NIR laser pulses (3 μs pulse width) with decreasing powers. The high stability of the nitrogen–vacancy emission enables us to define eight different output states. In this fashion, the nitrogen–vacancy centre gated by a NIR laser can be operated as a multi-level digital modulator. It is worth underlining that a single nitrogen–vacancy centre can be routinely used over several days without noticing any degradation in the emission properties. Furthermore, identical results were repeated on several nitrogen–vacancy centres from the same batch as well as on nitrogen–vacancy centres from different providers.
To assess the time response of the switch effect, the NIR pulse duration was changed from 3 μs to 200 ns while maintaining a fixed NIR laser power (Fig. 2d). The magnitude of the fluorescence drop is conserved for NIR pulses as short as 200 ns. We actually attribute this limit to the time response of the AOM used for the NIR laser modulation and expect the ultimate commutation rate to be only limited by the natural E→A decay rate of the nitrogen–vacancy.
The dynamics and response of the fluorescence from a gated single nitrogen–vacancy is thoroughly analysed in Fig. 3, where we find excellent agreement with a model based on a master rate equation involving just three energy bands (Fig. 3a): the excited and ground states A and E, as well as a dark state D that is reached on NIR illumination. After a few microseconds of continuous-wave green-laser illumination, the system evolves towards the ms = 0 sublevel of the ground triplet state manifold18, and thus, we can assume that states A and E are spin polarized, with a high probability into the ms = 0 levels of the ground and excited nitrogen–vacancy triplets. In fact, as most of the excitation events are associated with the intense and broad phonon sideband of the nitrogen–vacancy centre, narrow zero-phonon line features may be ignored in the fluorescence process. In what follows, we show that the NIR laser drives the system from E to A through an additional decay channel at very high rate compared with the natural decay rate κEA.

a, Model of energy bands for a nitrogen–vacancy centre gated with a NIR laser, consisting of a triplet ground state A, a triplet excited state E and a fast decaying dark band D. b, Saturation of the nitrogen–vacancy fluorescence as a function of green-laser power PG in the absence of a NIR laser (PNIR = 0). The red solid curve is a fit of the fluorescence F to the formula extracted from the three-level rate equation model sketched in a (see Supplementary Information): F = η κEA(α PG/(1+α PG+β PNIR)), where η is the detection efficiency, κEA is the natural E→A decay rate and α = 0.009 μW−1. c, Time-resolved measurements performed on a single nitrogen–vacancy centre after excitation with a 100 ps green laser under continuous NIR illumination. The measured power dependence of the excited-state lifetime (black dots) is extracted from the fits, as shown in the inset for three different NIR powers. The red solid curve is a fit of the fluorescence lifetime to the model, τE = (κEA)−1(1+β PNIR)−1, using β = 24.8 μW−1. d, Normalized drop in background-corrected fluorescence with increasing NIR laser power for different levels of continuous-wave green-laser excitation power (see labels), corresponding to the vertical lines in b with the same colour code. The solid curves are a fit of the fluorescence signal F using the previously fitted values of parameters α and β. All data are acquired from the same single nitrogen–vacancy centre, and all theory curves are calculated for the same values of parameters α and β.
In the absence of NIR illumination, only states A and E are involved. Excitation of the system from A to E occurs at a rate κAE = α PGκEA proportional to the green-laser power PG, whereas decay from E to A proceeds through radiative decay with a rate κEA = 1/(35 ns), which can change for different nitrogen–vacancy centres27. In our time-averaged measurements, it is safe to neglect quantum coherences and describe the system through a master rate equation, which leads to a fluorescence rate (see Supplementary Information) F = η κEAα PG/(1+α PG), where η = 0.005 is the detection efficiency of our system28. This produces an excellent fit to the green-laser-power-dependent fluorescence rate by choosing α = 0.009 μW−1 (Fig. 3b).
The effect of the NIR laser is best observed through the reduction that it produces in the lifetime of the excited state E (Fig. 3c). The nitrogen–vacancy is first prepared in the ms = 0 manifold on extended green-laser illumination followed by a 1 μs waiting time and is then excited with a 100 ps green-laser pulse. The ensuing time-resolved fluorescence reveals that the lifetime, which is controlled by both natural decay at a rate κEA and NIR-assisted decay at a rate β PNIRκAE, must thus follow the expression τE = (κEA)−1(1+β PNIR)−1. This produces an excellent fit to the measured data with the choice β = 24.8 μW−1 (Fig. 3c).
The full dependence of the fluorescence rate on green-laser and NIR-laser powers predicted by the rate-equation model reduces to (see Supplementary Information)

This simple expression is actually reproducing a detailed set of measured NIR-power-dependent profiles for different green-laser powers rather accurately with the single above-mentioned choice of parameters α and β (Fig. 3d), thus corroborating the validity of our model. The main assumption of the model is the additional fast decay proportional to PNIR, which is plausibly mediated by promotion to a dark band D. The nature of this band is an open question, although it is probably connected with higher-energy states reached through NIR photon absorption from the E band. Higher-energy states are more susceptible to decay non-radiatively, as they can access a larger number of levels through Auger and other electronic processes mediated by Coulomb interaction. In our model, we assume a promotion rate κED = β PNIR proportional to the NIR power, followed by fast decay from D to the ground state A. This is compatible with the sub-femtosecond timescale that is characteristic of non-radiative electronic transitions. Actually, the best fit to our data is obtained under the assumption κDA≫κEA (see Supplementary Information).
The fast, efficient modulation of the emission of a single nitrogen–vacancy centre presented here demonstrates the principle of a single-emitter optical switch operating at room temperature. A logical next step towards the exploitation of this concept for optical signal processing into an integrated scheme would consist of coupling a single nitrogen–vacancy to highly confined optical guided modes, as those supported for instance by a quasi-one-dimensional plasmonic waveguide29,30.
References
Knill, E., Laflamme, R. & Milburn, G. J. A scheme for efficient quantum computation with linear optics. Nature 409, 46–52 (2001).
Hwang, J. et al. A single-molecule optical transistor. Nature 460, 76–80 (2009).
Marklund, M. & Shukla, P. Nonlinear collective effects in photon–photon and photon–plasma interactions. Rev. Mod. Phys. 78, 591–640 (2006).
Hu, X., Jiang, P., Ding, C., Yang, H. & Gong, Q. Picosecond and low-power all-optical switching based on an organic photonic-bandgap microcavity. Nature Photon. 2, 185–189 (2008).
Albert, M., Dantan, A. & Drewsen, M. Cavity electromagnetically induced transparency and all-optical switching using ion Coulomb crystals. Nature Photon. 5, 633–636 (2011).
Fushman, I. et al. Controlled phase shifts with a single quantum dot. Science 320, 769–772 (2008).
Weis, S. et al. Optomechanically induced transparency. Science 330, 1520–1523 (2010).
Chang, D. E., Sørensen, A. S., Demler, E. A. & Lukin, M. D. A single- photon transistor using nanoscale surface plasmons. Nature Phys. 3, 807–812 (2007).
Orrit, M. Photons pushed together. Nature 460, 42–44 (2009).
Segal, M. Single-molecule transistor: Light switch. Nature Nanotech. 4, 472 (2009).
Moerner, W. E. Illuminating single molecules in condensed matter. Science 283, 1670–1676 (1999).
Wrigge, G., Gerhardt, I., Hwang, J., Zumofen, G. & Sandoghdar, V. Efficient coupling of photons to a single molecule and the observation of its resonance fluorescence. Nature Phys. 4, 60–66 (2007).
Mahler, B. et al. Towards non-blinking colloidal quantum dots. Nature Mater. 7, 659–664 (2008).
Chen, O. et al. Compact high-quality CdSe–CdS core–shell nanocrystals with narrow emission linewidths and suppressed blinking. Nature Mater. 12, 445–451 (2013).
Doherty, M. W. et al. The nitrogen–vacancy colour centre in diamond. Phys. Rep. 528, 1–45 (2013).
Cai, J., Retzker, A., Jelezko, F. & Plenio, M. B. A large-scale quantum simulator on a diamond surface at room temperature. Nature Phys. 9, 168–173 (2013).
Balasubramanian, G. et al. Ultralong spin coherence time in isotopically engineered diamond. Nature Mater. 8, 383–387 (2009).
Jelezko, F. & Wrachtrup, J. Single defect centres in diamond: A review. Phys. Status Solidi a 203, 3207–3225 (2006).
Neumann, P. et al. Excited-state spectroscopy of single NV defects in diamond using optically detected magnetic resonance. New J. Phys. 11, 013017 (2009).
Delaney, P., Greer, J. C. & Larsson, J. A. Spin-polarization mechanisms of the nitrogen-vacancy center in diamond. Nano Lett. 10, 610–614 (2010).
Irie, M., Kobatake, S. & Horichi, M. Reversible surface morphology changes of a photochromic diarylethene single crystal by photoirradiation. Science 291, 1769–72 (2001).
Pärs, M. et al. An organic optical transistor operated under ambient conditions. Angew. Chem. Int. Ed. 50, 11405–8 (2011).
Rittweger, E., Han, K. Y., Irvine, S. E., Eggeling, C. & Hell, S. W. STED microscopy reveals crystal colour centres with nanometric resolution. Nature Photon. 3, 144–147 (2009).
Lai, N. D. et al. Quenching nitrogen–vacancy center photoluminescence with an infrared pulsed laser. New J. Phys. 15, 033030 (2013).
Toyli, D. M. et al. Measurement and control of single nitrogen–vacancy center spins above 600 K. Phys. Rev. X 2, 031001 (2012).
Neukirch, L. P., Gieseler, J., Quidant, R., Novotny, L. & Vamivakas, A. N. Observation of nitrogen vacancy photoluminescence from an optically levitated nanodiamond. Opt. Lett. 38, 2976–2979 (2013).
Tisler, J. et al. Fluorescence and spin properties of defects in single digit nanodiamonds. ACS Nano. 3, 1959–1965 (2009).
Brouri, R., Beveratos, A., Poizat, J-P. & Grangier, P. Photon antibunching in the fluorescence of individual color centers in diamond. Opt. Lett. 25, 1294–1296 (2000).
Bozhevolnyi, S. I., Volkov, V. S., Devaux, E., Laluet, J-Y. & Ebbesen, T. W. Channel plasmon subwavelength waveguide components including interferometers and ring resonators. Nature 440, 508–511 (2006).
Gonzalez-Tudela, A., Martin-Cano, D., Moreno, E., Martin-Moreno, L., Tejedor, C. & Garcia-Vidal, F. J. Entanglement of two qubits mediated by one-dimensional plasmonic waveguides. Phys. Rev. Lett. 106, 20501 (2011).
Acknowledgements
This work was partially supported by the Spanish Ministry of Sciences (grants FIS2010– 14834, MAT2010-14885 and CSD2007–046-NanoLight.es), the European Community’s Seventh Framework Program under grant ERC-Plasmolight (no. 259196) and Fundació privada CELLEX. M.G. acknowledges the support of the FPU grant AP2009-3025 from the Spanish Ministry of Education. R.M. acknowledges support of Marie Curie and NEST programmes. The authors thank M. Orrit for fruitful discussions.
Author information
Authors and Affiliations
Contributions
M.G. and R.Q. conceived the experiment. M.G. and R.M. performed the experiments, F.J.G.d.A. provided theoretical support. All authors discussed the results and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1233 kb)
Rights and permissions
About this article
Cite this article
Geiselmann, M., Marty, R., García de Abajo, F. et al. Fast optical modulation of the fluorescence from a single nitrogen–vacancy centre. Nature Phys 9, 785–789 (2013). https://doi.org/10.1038/nphys2770
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys2770
This article is cited by
-
Tuning colour centres at a twisted hexagonal boron nitride interface
Nature Materials (2022)
-
Dynamic upconversion multicolour editing enabled by molecule-assisted opto-electrochemical modulation
Nature Communications (2021)
-
Fast electrical modulation of strong near-field interactions between erbium emitters and graphene
Nature Communications (2020)
-
Modification of single molecule fluorescence using external fields
Frontiers of Physics (2017)
-
Electron spin control of optically levitated nanodiamonds in vacuum
Nature Communications (2016)
