
Abstract
Hexanucleotide repeat expansions represent the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia, though the mechanisms by which such expansions cause neurodegeneration are poorly understood. We report elevated levels of DNA–RNA hybrids (R-loops) and double strand breaks in rat neurons, human cells and C9orf72 ALS patient spinal cord tissues. Accumulation of endogenous DNA damage is concomitant with defective ATM-mediated DNA repair signaling and accumulation of protein-linked DNA breaks. We reveal that defective ATM-mediated DNA repair is a consequence of P62 accumulation, which impairs H2A ubiquitylation and perturbs ATM signaling. Virus-mediated expression of C9orf72-related RNA and dipeptide repeats in the mouse central nervous system increases double strand breaks and ATM defects and triggers neurodegeneration. These findings identify R-loops, double strand breaks and defective ATM-mediated repair as pathological consequences of C9orf72 expansions and suggest that C9orf72-linked neurodegeneration is driven at least partly by genomic instability.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Schottlaender, L.V. et al. Analysis of C9orf72 repeat expansions in a large series of clinically and pathologically diagnosed cases with atypical Parkinsonism. Neurobiol. Aging 36, 1221.e1–1221.e6 (2015).
Rutherford, N.J. et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 33, 2950.e5–2950.e7 (2012).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Groh, M. & Gromak, N. Out of balance: R-loops in human disease. PLoS Genet. 10, e1004630 (2014).
Roy, D., Yu, K. & Lieber, M.R. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol. Cell. Biol. 28, 50–60 (2008).
Aguilera, A. & García-Muse, T. R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124 (2012).
Haeusler, A.R. et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014).
Bhatia, V. et al. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511, 362–365 (2014).
Yüce, Ö. & West, S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 33, 406–417 (2012).
Lindquist, S.G. et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin. Genet. 83, 279–283 (2013).
Corcia, P. et al. Pure cerebellar ataxia linked to large C9orf72 repeat expansion. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 301–303 (2016).
McKinnon, P.J. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 7, 303–321 (2012).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013).
Frappart, P.-O. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 11, 538–544 (2005).
Ashour, M.E., Atteya, R. & El-Khamisy, S.F. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat. Rev. Cancer 15, 137–151 (2015).
Gómez-Herreros, F. et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 46, 516–521 (2014).
Chiang, S.-C. et al. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci. Adv. 3, e1602506–e1602516 (2017).
Harrigan, J.A. et al. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 (2011).
Heo, J.-I. et al. ATM mediates interdependent activation of P53 and ERK through formation of a ternary complex with p-P53 and p-ERK in response to DNA damage. Mol. Biol. Rep. 39, 8007–8014 (2012).
Katyal, S. et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci. 17, 813–821 (2014).
Alagoz, M., Chiang, S.-C., Sharma, A. & El-Khamisy, S.F. ATM deficiency results in accumulation of DNA-topoisomerase I covalent intermediates in neural cells. PLoS One 8, e58239 (2013).
Li, J. et al. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 18, 783–790 (2012).
Zhang, Y.-J. et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 19, 668–677 (2016).
Difilippantonio, S. et al. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 7, 675–685 (2005).
Schwertman, P., Bekker-Jensen, S. & Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 17, 379–394 (2016).
Shanbhag, N.M., Rafalska-Metcalf, I.U., Balane-Bolivar, C., Janicki, S.M. & Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981 (2010).
Bohgaki, M. et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 110, 20982–20987 (2013).
Fradet-Turcotte, A. et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54 (2013).
Doil, C. et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446 (2009).
Noon, A.T. et al. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 12, 177–184 (2010).
Lee, J.-H., Goodarzi, A.A., Jeggo, P.A. & Paull, T.T. 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 29, 574–585 (2010).
Stewart, G.S. et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434 (2009).
Wang, Y. et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/P62. Mol. Cell 63, 34–48 (2016).
Mann, D.M.A. et al. Dipeptide repeat proteins are present in the P62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1, 68 (2013).
Davidson, Y.S. et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2, 70 (2014).
Tresini, M., Marteijn, J.A. & Vermeulen, W. Bidirectional coupling of splicing and ATM signaling in response to transcription-blocking DNA damage. RNA Biol. 13, 272–278 (2016).
Tresini, M. et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58 (2015).
Bakkenist, C.J. & Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003).
Belzil, V.V. et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905 (2013).
Goodarzi, A.A. et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177 (2008).
Meisenberg, C. et al. Epigenetic changes in histone acetylation underpin resistance to the topoisomerase I inhibitor irinotecan. Nucleic Acids Res. 45, 1159–1176 (2017).
Felisbino, M.B., Gatti, M.S.V. & Mello, M.L.S. Changes in chromatin structure in NIH 3T3 cells induced by valproic acid and trichostatin A. J. Cell. Biochem. 115, 1937–1947 (2014).
Sanz, L.A. et al. Prevalent, dynamic, and conserved R-loop structures associate with specific epigenomic signatures in mammal. Mol. Cell 63, 167–178 (2016).
Castellano-Pozo, M. et al. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 52, 583–590 (2013).
Zhang, Y. et al. Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat. Res. 168, 115–124 (2007).
Wang, X. et al. Alzheimer disease and amyotrophic lateral sclerosis: an etiopathogenic connection. Acta Neuropathol. 127, 243–256 (2014).
El-Khamisy, S.F. et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434, 108–113 (2005).
El-Khamisy, S.F. To live or to die: a matter of processing damaged DNA termini in neurons. EMBO Mol. Med. 3, 78–88 (2011).
Chan, Y.A. et al. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 10, e1004288 (2014).
Zu, T. et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 110, E4968–E4977 (2013).
McIntyre, G.J., Groneman, J.L., Tran, A. & Applegate, T.L. An infinitely expandable cloning strategy plus repeat-proof PCR for working with multiple shRNA. PLoS One 3, e3827 (2008).
Cooper-Knock, J. et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137, 2040–2051 (2014).
Mead, R.J. et al. Optimised and rapid pre-clinical screening in the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis (ALS). PLoS One 6, e23244 (2011).
McCloy, R.A. et al. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400–1412 (2014).
Acknowledgements
We thank A. Isaacs for providing the original DPR plasmids, G. Stewart for providing the RNF168 plasmid and L. Baxter for the immunohistochemistry of postmortem sections. M.A. and S.F.E.-K. made equal financial contributions to the work through the European Research Council Award (294745) and the Wellcome Trust Investigator Award (103844), respectively. S.F.E.-K. is funded by a Wellcome Trust Investigator Award (103844), a Lister Institute of Preventative Medicine Fellowship and a European Union British Council award. M.A. is funded by the European Research Council (ERC Advanced Award no. 294745) and MRC DPFS Award (129016/G1001492). C.L., S.-C.C., M.J., W.E., S.R. and C.W. (50%) were supported by the Wellcome Trust Investigator Award (103844). E.K. was supported by an Eve Davis Studentship. S.H.-M., I.T., V.L., K.L., J.C. and C.W. (50%) were supported by the ERC Advanced Award (294745). P.J.S. and G.M.H. are supported by the Medical Research Council (MR/M010864/1) and P.J.S. is supported as an NIHR Senior Investigator. K.J.D.V. is supported by the Thierry Latran Foundation (Project RoCIP) and Medical Research Council (MR/K005146/1 and MR/M013251/1).
Author information
Authors and Affiliations
Contributions
C.W. conducted the immunocytochemistry, immunohistochemistry and immunoblotting experiments. S.H.-M. generated the mouse model and conducted behavioral experiments with assistance from K.L., V.L., T.I., J.C. and I.C. E.K. and S.H.-M. generated neuronal cell cultures. E.K., S.-C.C., M.J., S.R. and M.K.H. assisted with cell culture, imaging and DNA repair assays. K.L., V.L., T.I. and I.C. assisted with mouse experiments. C.L. purified and conducted TOP1cc assays. K.J.D.V., I.T., J.C. and A.H. generated expansion constructs. I.T. and S.H.-M. generated and validated viral vector stocks. W.E. optimized and conducted the neutral comet assays. P.J.S. and A.H. provided RRE constructs and I.T., J.C. and P.J.M. subcloned them into viral vectors. P.J.S. and A.H. provided the C9orf72 ALS biosamples and expertise. S.F.E.-K., M.A., G.M.H., C.W. and S.H.-M. analyzed the data. S.F.E.-K. and C.W. wrote the manuscript with help from M.A. and S.H.-M. All authors contributed to the final manuscript. S.F.E.-K. and M.A. conceived and co-led the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Expression of GFP or V5 empty vector does not lead to defective ATM phosphorylation in MRC5 cells
(a) MRC5 cells mock transfected, or transfected with constructs encoding a 0-V5 empty vector or GFP, were immunostained with anti-phosphorylated ATM ‘pATM’ antibodies. Endogenous GFP signal was visualised without antibody amplification. Nuclei were counterstained with DAPI, and representative images of 2 biological replicates are shown, scale bar 5μm. (b) The number of pATM foci per cell was quantified and presented as average ± range from 2 independent cell culture replicates, 50 cells each.
Supplementary Figure 2 ATM inhibition, but not the expression of GFP or V5 empty vectors, suppresses 53BP1 foci formation
(a) Left, MRC5 cells mock transfected or transfected with 0-V5 or GFP were immunostained with anti-53BP1 antibodies. Endogenous GFP signal was visualised without antibody amplification. Nuclei were counterstained with DAPI, and representative images of 2 biological replicates are shown, scale bar 5μm. Right, the number of 53BP1 foci per cell was quantified and presented as average ± range from 2 biological replicates, 50 cells each. (b) Left, Rat cortical neurons mock transduced, or transduced with AAV9 encoding a V5 epitope-tag or GFP, were incubated with 10μM CPT for 1 hour. Subsequently, cells were immunostained with anti-53BP1 antibodies. Nuclei were counterstained with DAPI and representative images of 2 biological replicates are shown, scale bar 5μm. Right, The number of 53BP1 foci per cell was quantified and presented as average ± range from 2 biological replicates, 20 cells each. (c) MRC5 cells treated with Ku5593 (10μM) for 2 hours were analysed by immunocytochemistry using phospho-ATM antibodies. The number of pATM nuclear foci was quantified from 3 biological replicates, 25 cells each, and is presented the average ± SEM. Significance assessed using a Student’s t-test. (d,e) MRC5 cells transfected with 0, 34, or 69 poly-GA DPRs, and cells were treated with Ku5593 (10μM) for 2 hours. Subsequently, cells were analysed by immunocytochemistry using anti-53BP1 (d) or anti-phospho-p53 (e) antibodies. The number of 53BP1 (b) or p-p53 (e) nuclear foci was quantified from 3 biological replicates, 25 cells each, and is presented the average ± SEM. Significance assessed using a Student’s t-test.
Supplementary Figure 3 Expression of C9orf72 poly-GA DPRs in rat cortical neurons leads to the nuclear enrichment of HDAC4
(a) Rat cortical neurons were mock transduced or transduced with a AAV9 viral vectors encoding 69 DPRs, and analysed by immunocytochemistry using anti-V5 ‘DPRs’ and an anti-HDAC4 antibodies. Nuclei were counterstained with DAPI and representative images are shown, scale bar 5μm. (b) The % of cultured cortical neurons exhibiting nuclear HDAC4 staining was quantified from 3 neuronal preparation replicates, 20 neurons each, and presented as average ± SEM. Significance was assessed using a Student’s t-test. (c) Rat cortical neurons were mock transduced or transduced with V5-epitope tagged poly-GA 69 DPRs, and analysed by immunocytochemsitry using anti-V5 and anti-MAP2 antibodies. Nuclei were counterstained with DAPI and representative images of 3 neuronal preparation replicates are shown, scale bar 5μm. (d) Rat cortical neurons were mock transduced or transduced with AAV9 viral particles encoding a V5 epitope-tag or GFP. Neurons were treated with an ATM inhibitor ‘ATMi’ (Ku 55933, 10μM) for 1 hour, and subsequently were immunostained with anti-HDAC4 antibodies. Nuclei were counterstained with DAPI and representative images of 2 neuronal preparation replicates are shown, scale bar 5μm. (e) The percentage of cells with cytoplasmic HDAC4 was quantified and presented as average ± range from 2 biological replicates, 20 cells each.
Supplementary Figure 4 Cisterna magna injection of scAAV9 viral particles encoding 69 DPRs leads to DPR aggregates in the CNS
Brain sections (40μM thickness) from mice aged 1 month, were subjected to immunohistochemical analyses using anti-V5 (Bethyl, A190-120A) and anti-NeuN (Merck Millipore,MAB377) antibodies. Nuclei were counterstained with Hoechst and representative images from 3 animals are shown, scale bar 20μM and 5μM for unzoomed and zoomed images, respectively.
Supplementary Figure 5 Mice injected with 69 DPRs display further behavioral deficits at age 12 months, in comparison to 0-DPR-injected controls
Mice injected with 0 or 69 DPRs were assayed for splay differences, aged 12 months, using neuroscoring analysis. Neuroscores are presented as average (n=8 and n=9 for 0-V5 and 69-V5, respectively) ± SEM. Significance was assessed using a Student’s t-test.
Supplementary Figure 6 Chloroquine increases ATM activation and restores cytoplasmic HDAC4 localisation in DPR-expressing cells
(a) Left, MRC5 cells transfected with constructs encoding 0, 69 poly-GADPRs were treated with chloroquine (320μg/ml) ‘+CQ’ or water ‘-CQ’ for 4 hours. Subsequently, cells were immunostained with anti-V5 ‘DPRs’ and anti-ATM ‘pATM’ antibodies. Nuclei were counterstained with DAPI and representative images of 3 cell culture replicates are shown, scale bar 5μm. Right, Nuclear fluorescence intensity was quantified and data presented as the average ± SEM from 3 biological repeats, 25 cells each. Significance was assessed using a Student’s t-test. (b) Left, Rat cortical neurons were transduced with AAV9-viral vectors encoding 0 or 69 DPRs, and analysed by immunocytochemistry using anti V5 ‘DPRs’ and an anti-HDAC4 antibodies. Nuclei were counterstained with DAPI and representative images are shown, scale bar 5μm. Right, The % of cultured cortical neurons exhibiting cytoplasmic HDAC4 staining was quantified from 3 neuronal preparation replicates, 20 neurons each, and presented as average ± SEM. Significance was assessed using a Student’s t-test.
Supplementary Figure 7 C9orf72-associated DSBs and cellular toxicity can be reduced by chromatin decondensation
(a) Left, HEK 293T cells transfected with constructs encoding 0 or 69 DPRs were analysed using Western Blotting with anti-H3K9me3 and anti-GAPDH antibodies. Right, H3K9me3 band intensity quantified and normalised to GAPDH, data presented as the average ± SEM from 4 cell culture replicates and analysed using Student’s t-test. (b) Left, MRC5 cells transfected with 0 or 69 DPRs were immunostained with anti-V5 ‘DPRs’ and H3K9me3 antibodies. Representative images of 3 independent cell culture replicates are shown. Right, MRC5 cells transfected with 0, 69 DPRs were treated with TSA (100nm) or DMSO and immunostained with anti-V5 ‘DPRs’ and H3K9me3 antibodies. Nuclear fluorescence intensity was quantified and presented as the average ± SEM from 3 independent cell culture repeats, 25 cells each. Significance was assessed using a Student’s t-test. (c) MRC5 transfected 0 or 69 GA DPRs were treated with TSA or DMSO and were immunostained with anti-V5 ‘DPRs’ and phosphorylated-ATM ‘pATM’ antibodies. Nuclear fluorescence intensity was quantified and presented as the average ± range from 2 independent cell culture replicates, 25 cells each. (d) MRC5 cells transfected with 0 or 69 DPRs and were treated with TSA or DMSO. Cells were immunostained with anti-V5 ‘DPRs’ and anti γH2AX antibodies. The average (± SEM) percentage of cells with 10 or more γH2AX foci was quantified from 3 independent cell culture replicates, 50 cells each, and analysed using a Student’s t-test. (e) MRC5 cells transfected with 0 or 69 DPRs were treated with TSA or DMSO, and immunostained with anti-V5 ‘DPRs’ and anti S9.6 antibodies. The average (± range) number of R-Loop foci per cell was quantified from 2 independent cellular replicates, 25 cells each. (f) MRC5 cells transfected with 0 or 69 DPRs and were treated with TSA or DMSO, and the average (± SEM) percentage of cells positive for cleaved PARP was quantified from 3 independent cell culture replicates, 50 cells each, and analysed using a Student‘s t-test. (g) HEK 293T cells transfected with 0 or 69 DPRs were treated with TSA or DMSO. The average (± SEM) percentage of cells permeable to Trypan Blue reagent was quantified from 5 independent replicates, ~200 cells each, and was analysed using a Student’s t-test. (h) MRC5 cells transfected with 0 or 69 DPRs. Prior to transfection, cells were transduced with adenoviral vectors encoding for RFP or SETX. Subsequently, cells were immunostained with anti-V5 ‘DPRs’ and anti-H3K9me3 antibodies. Nuclear fluorescence intensity was quantified and data presented as the average ± SEM from 3 independent cell culture repeats, 25 cells each. Significance was assessed using a Student’s t-test. (i) MRC5 cells transduced with adenoviral vectors encoding for SETX or RFP were transfected with constructs encoding 0 or 69 DPRs, and then treated with TSA or DMSO. Cells were immunostained with anti-V5 ‘DPRs’ and anti--γH2AX antibodies. Nuclei were counterstained with DAPI. The percentage of cells with 10 or more γH2AX foci was quantified and presented as average ± SEM from 2 independent replicates, 25 cells each. (j) MRC5 cells transfected with 0 or 69 dipeptides, alongside control or p62 siRNA particles, were treated with TSA or DMSO. Subsequently, cells were immunostained with anti-V5 ‘DPRs’ and anti-H3K9me3 antibodies. Nuclear fluorescence intensity was quantified and data presented as the average ± SEM from 3 independent cell culture repeats, 25 cells each. Significance was assessed using a Student’s t-test. (k) MRC5 cells transfected with 0 or 69 DPRs, alongside control or p62 siRNA particles, were treated with TSA or DMSO. Cells were immunostained with anti-V5 ‘DPRs’ and anti--γH2AX antibodies. Nuclei were counterstained with DAPI. The percentage of cells with 10 or more γH2AX foci was quantified and presented as average ± SEM from 2 independent cell culture replicates, 25 cells each.
Supplementary Figure 8 RNASE H1 preincubation ablates S9.6 signal in postmortem sections
Human post-mortem sections were pretreated with RNAse H (25U/ml) NEB, M0297S) overnight at 4°C or were left untreated. Subsequently, sections were subjected to immunohistochemical analysis with the R-Loop specific, phage developed, S9.6 antibodies. Representative images from the selected antibody optimisation conditions are presented, scale bar 250μm.
Supplementary Figure 9 Spinal cord motor neurons from C9orf72 ALS postmortem show elevated levels of heterochromatin
Left, Human spinal cord sections were analysed by immunohistochemistry using anti-H3K9me3 (Abcam, ab8898) antibodies. Representative images from 4 C9orf72 patient and 4 control sections are presented, scale bar 10μm. Right, The mean nuclear H3K9me3 fluorescence of motor neurons was quantified from 4 C9orf72 patient and 4 control sections and is presented as average ± SEM, with ~20 cells each. Significance was assessed using a Student’s t-test.
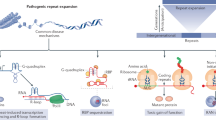
Supplementary Figure 10 Model describing how C9orf72 expansions cause genomic instability
Transcription of C9orf72 repeat expansions causes elevated R-loop levels, leading to chromosomal breaks. Concomitantly, C9orf72 expansions cause the hallmark accumulation of p62, which inhibits H2A ubiquitination, and disrupts ATM-signaling. Defective ATM signaling prevents 53BP1-mediated DNA repair, exacerbating the genomic instability driven by R-loops. R-loops and defective ATM signaling are distinguishable, yet interlinked, pathways that contribute to genome instability, leading to the disruption of co-transcriptional processing, and, ultimately, neuronal cell death in ALS/FTD.
Supplementary Figure 11 Left: full uncropped western blot image from Figure 1l. Right: full uncropped western blot image from Figure 1n.
Dashed box represents the cropped image used in main figure.
Supplementary Figure 12 Full uncropped western blot image from Figure 2c.
Dashed box represents the cropped image used in main figure.
Band below tubulin represents GAPDH immunoblot at 37kDa (which did not work well as was at the bottom of the gel).
Supplementary Figure 13 Top: full uncropped western blot image from Figure 4e. Bottom: full uncropped western blot from Figure 5b.
Dashed box represents the cropped image used in main figure.
Supplementary Figure 14 Full uncropped western blot image from Figure 5f and Supplementary Figure 7a.
Dashed box represents the cropped image used in figure.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–14 and Supplementary Table 1. (PDF 1763 kb)
Rights and permissions
About this article

Cite this article
Walker, C., Herranz-Martin, S., Karyka, E. et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci 20, 1225–1235 (2017). https://doi.org/10.1038/nn.4604
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.4604
This article is cited by
-
Chromosomal R-loops: who R they?
Biologia Futura (2024)
-
Molecular hallmarks of ageing in amyotrophic lateral sclerosis
Cellular and Molecular Life Sciences (2024)
-
Neuronal STING activation in amyotrophic lateral sclerosis and frontotemporal dementia
Acta Neuropathologica (2024)
-
Small molecule modulators of chromatin remodeling: from neurodevelopment to neurodegeneration
Cell & Bioscience (2023)
-
A distinct circular DNA profile intersects with proteome changes in the genotoxic stress-related hSOD1G93A model of ALS
Cell & Bioscience (2023)
