
Abstract
SGK1 is an AGC kinase that regulates the expression of membrane sodium channels in renal tubular cells in a manner dependent on the metabolic checkpoint kinase complex mTORC2. We hypothesized that SGK1 might represent an additional mTORC2-dependent regulator of the differentiation and function of T cells. Here we found that after activation by mTORC2, SGK1 promoted T helper type 2 (TH2) differentiation by negatively regulating degradation of the transcription factor JunB mediated by the E3 ligase Nedd4-2. Simultaneously, SGK1 repressed the production of interferon-γ (IFN-γ) by controlling expression of the long isoform of the transcription factor TCF-1. Consistent with those findings, mice with selective deletion of SGK1 in T cells were resistant to experimentally induced asthma, generated substantial IFN-γ in response to viral infection and more readily rejected tumors.
Similar content being viewed by others


Main
AGC kinases are serine-threonine kinases that are broadly involved in regulating various aspects of cell physiology, including growth, survival and metabolism1. SGK1 ('serum- and glucocorticoid-regulated kinase 1') is an AGC kinase that is activated by sequential phosphorylation of two highly conserved regulatory motifs: the T-loop domain is phosphorylated at Thr256 by the kinase PDK1 (ref. 2), and the hydrophobic motif is phosphorylated at Ser422 by the metabolic checkpoint kinase complex mTORC2 (refs. 3,4). Compared with other AGC kinases that are also downstream of mTOR, such as Akt and S6 kinase, relatively little is known about SGK1. Downstream targets of SGK1 include E3 ubiquitin ligases such as Nedd4-2 (refs. 5,6), transcription factors such as Foxo7,8 and other kinases such as GSK-3β9. In the kidney, SGK1 phosphorylates and inhibits Nedd4-2 to prevent degradation of the epithelial sodium channel and thus allows sodium to be reclaimed in the urine when serum concentrations of sodium are low5,10,11. It has been shown that SGK1 mRNA is upregulated by sodium in lymphocytes and that mice fed a high-salt diet develop more severe autoimmune encephalomyelitis mediated by the TH17 subset of helper T cells12,13. Loss of SGK1 in T cells leads to a selective defect in pathogenic TH17 differentiation due to decreased expression of the receptor for interleukin 23 (IL-23)12. The role of SGK1 in other T cell lineages, however, is yet to be determined.
We became interested in SGK1 because it is a downstream target of mTOR, which serves as a critical node in a highly conserved signaling pathway that integrates multiple inputs from the environment14,15. In the immune system, mTOR integrates various signals, such as cytokines and costimulatory molecules, to influence T cell differentiation15. mTOR can associate with two distinct protein complexes (mTORC1 and mTORC2) to drive the selective differentiation of CD4+ T cells. Deletion of the gene encoding mTOR in CD4+ T cells has shown that loss of signaling via both mTORC1 and mTORC2 results in differentiation toward a 'default' regulatory T cell phenotype upon activation of T cells16,17. Loss of either mTORC1 or mTORC2 leads to selective deficits in distinct T cell lineages. For example, deletion of the gene encoding the mTORC2 adaptor Rictor in CD4+ T cells results in defective T helper type 2 (TH2) differentiation characterized by an inability to produce IL-4 (refs. 18,19). Despite the critical role of mTOR in regulating the differentiation of effector T cells and regulatory T cells, the precise downstream targets of mTOR that control differentiation into distinct helper T cell subsets have yet to be elucidated.
Because SGK1 is a downstream target of mTORC2, we hypothesized that it might be involved in regulating commitment to the TH2 lineage by mTORC2. To investigate this, we crossed mice with loxP-flanked alleles encoding (Sgk1fl/fl)11 with mice expressing Cre recombinase from the Cd4 promoter to generate 'T-Sgk1−/−' progeny in which SGK1 was selectively deleted in T cells. We found that loss of SGK1 led to less production of IL-4 and inappropriate release of interferon-γ (IFN-γ) under TH2-polarizing conditions in vitro. In a mouse model of allergic asthma, we found that loss of SGK1 resulted in diminished TH2 responses and protection against disease. Mechanistically, we found that SGK1 promoted TH2 differentiation by preventing ubiquitination and degradation of the transcription factor JunB by Nedd4-2 and its adaptor Ndfip. Alternatively, we found that T-Sgk1−/− mice produced more IFN-γ under TH1-polarizing conditions in vivo than did wild-type mice, and this was associated with stronger antitumor and antiviral immune responses. The ability of SGK1 to inhibit TH1 differentiation was mediated in part by its ability to regulate expression of the long isoform of the transcription factor TCF-1 via β-catenin and GSK-3β. Overall, our findings define distinct signaling pathways downstream of SGK1 that provide new evidence about how mTOR controls helper T cell differentiation.
Results
Activation of SGK1 after recognition of antigen in T cells
SGK1 RNA was expressed in resting naive T cells (Supplementary Fig. 1a). We wanted to determine whether SGK1 was activated upon stimulation of T cells by measuring phosphorylation of the SGK1 substrate NDRG1 ('N-myc-downregulated gene 1') at Thr346 (ref. 20). Activation of T cells led to an increase in SGK1 activity (Fig. 1a). The kinetics of SGK1 activation paralleled those of the mTORC2-dependent activation of Akt (phosphorylation of Akt at Ser473) (Fig. 1a).

(a) Immunoblot analysis of NDRG1 phosphorylated at Thr346 (p-NDRG1(T346)) (SGK1 activity) and Akt phosphorylated at Ser473 (p-Akt(S473)) (mTORC2 activity) in lysates of naive lymphocytes obtained from 5C.C7 mice and allowed to 'rest' for 1 h before stimulation for 0, 1 or 3 h (above lanes) with antibody to CD3 (anti-CD3) and anti-CD28 (TCR + costim); total protein (NDRG1 and Akt) and actin serve as loading controls (throughout). (b) Immunoblot analysis (as in a) of lysates of CD4+ T cells isolated from 5C.C7 mice as in a and stimulated for 3 h in the presence of polarizing cytokines (above lanes). (c) Immunoblot analysis (as in a) of naive 5C.C7 lymphocytes treated as in a, with (+) or without (−) addition of the mTOR-kinase inhibitor PP242 (1 μM) during stimulation. Data are representative of three to five independent experiments.
Next we investigated whether SGK1 activity was modulated by polarizing cytokines upon T cell activation. Consistent with a published report12, SGK1 mRNA was expressed under all polarizing conditions in vitro, but its expression was highest under TH17-polarizing conditions (Supplementary Fig. 1b). Notably, we found that SGK1 activity was induced to a similar degree under all polarizing conditions (Fig. 1b).
Given the role of mTOR in regulating SGK1 activity in HEK293T human embryonic kidney cells, MCF-7 human breast cancer cells and HeLa human cervical cancer cells3,21, we sought to determine whether SGK1 was also activated in an mTOR-dependent manner in T cells (Supplementary Fig. 2a). Phosphorylation of NDRG1 increased after T cells were stimulated, but that effect was abrogated in the presence of PP242, an inhibitor of the kinase activity of mTOR22 (Fig. 1c). Consistent with those results, we also observed inhibition of the phosphorylation of Akt at Ser473 (Fig. 1c), which indicated successful blockade of mTORC2 activity23,24,25. Likewise, T cell–specific deletion of the mTORC2 component Rictor through deletion of loxP-flanked Rictor alleles by Cd4 promoter–driven Cre (T-Rictor−/−) abrogated the activation of both Akt and SGK1 (Fig. 2a and Supplementary Fig. 2b,c).

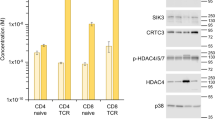
(a) Immunoblot analysis (as in Fig. 1a) of lysates of wild-type (WT), T-Rictor−/− and T-Sgk1−/− CD4+ T cells and allowed to 'rest' for 1 h before stimulation for 0 h or 3 h with anti-CD3 and anti-CD28. (b–e) Flow cytometry of cells from the spleen (b,c) and lymph nodes (d,e) of wild-type and T-Sgk1−/− mice (n = 6 per genotype), assessing the frequency of B220+ or CD3+ cells (b,d) and CD4+ or CD8+ cells (gated on CD3+ cells; c,e). Each symbol represents an individual mouse; small horizontal lines indicate the mean (±s.e.m.). (f) Flow cytometry of wild-type and T-Sgk1−/− CD4+ T cells stained with the division-tracking dye CFSE and left unstimulated (US) or stimulated (Stim) for 24, 48, 72 or 96 h (above plots) with irradiated syngeneic antigen-presenting cells and anti-CD3 (1 μg/ml); bottom row, overlay of plots of stimulated cells above. (g) Enzyme-linked immunosorbent assay (ELISA) of IL-2 in supernatants of naive CD4+ T cells stimulated overnight with plate-bound anti-CD3 (0.3, 1.0, or 3.0 μg/ml; horizontal axis) and soluble anti-CD28 (2.0 μg/ml). ND, not detectable. NS, not significant (Student's t-test). Data are representative of three independent experiments (error bars (g), s.e.m. of triplicates).
Having established that signaling via SGK1 was dependent on mTORC2 in T cells, we wanted to determine if SGK1 might function downstream of mTORC2 to regulate the differentiation of helper T cells. To address this issue, we bred Sgk1fl/fl mice11 with mice with Cd4 promoter–driven Cre expression to generate 'T-Sgk1−/−' progeny with selective deletion of SGK1 in T cells (Supplementary Fig. 1a). After stimulation, phosphorylation of NDRG1 was much lower in T-Sgk1−/− CD4+ T cells than in their wild-type counterparts (Fig. 2a). In contrast, selective deletion of SGK1 in T cells did not affect the phosphorylation of Akt or S6 (Fig. 2a and Supplementary Fig. 2b,c).
The role of SGK1 in the maturation of peripheral T cells has not been identified, to our knowledge. Thymic development was essentially not different in wild-type mice versus T-Sgk1−/− mice (Supplementary Fig. 3a), and there were no differences between wild-type and T-Sgk1−/− mice in the frequency of B220+, CD3+, CD4+ or CD8+ cells in the spleen and lymph nodes (Fig. 2b–e). We observed subtle differences between wild-type and T-Sgk1−/− mice in the absolute number of T cells and expression of activation markers (Supplementary Fig. 3b–g). Finally, the spleens of T-Sgk1−/− mice had a slightly greater abundance of Foxp3+ T cells (7.50%) than did those of wild-type mice (6.23%); however, we did not observe this difference in the lymph nodes (Supplementary Fig. 3h). T-Sgk1−/− CD4+ T cells displayed a slightly lower rate of proliferation than that of wild-type CD4+ T cells (Fig. 2f). Nevertheless, wild-type and T-Sgk1−/− CD4+ T cells produced similar amounts of IL-2 (Fig. 2g). Thus, the lack of SGK1 did not have a substantial effect on the composition of the peripheral T cell compartment.
SGK1 regulates TH1 and TH2 differentiation
Given the ability of mTORC2 to regulate the generation of TH2 cells18,19, we wanted to determine whether loss of SGK1 affected TH1 and TH2 differentiation. For this, we obtained CD4+ T cells from wild-type and T-Sgk1−/− mice, cultured the cells under nonpolarizing (TH0) or TH1- or TH2-polarizing conditions and assessed IL-4 production. As expected, wild-type T cells stimulated under TH2-polarizing conditions and rechallenged with anti-CD3 and anti-CD28 produced IL-4 (Fig. 3a). However, T-Sgk1−/− T cells did not produce substantial amounts of IL-4 under the same conditions (Fig. 3a). Furthermore, lack of SGK1 resulted in lower production of the TH2 cytokines IL-5 and IL-13 (Fig. 3b,c).

(a) ELISA of IL-4 in supernatants of naive wild-type and T-Sgk1−/−CD4+ T cells isolated on the basis of expression of CD44 and CD62L and then stimulated with irradiated autologous antigen-presenting cells under TH0 or TH1- or TH2-polarizing conditions (key), then allowed to 'rest' for 5 d before being restimulated with anti-CD3 and anti-CD28. (b,c) ELISA of IL-5 (b) or IL-13 (c) in supernatants of cells as in a. (d) Intracellular staining of IFN-γ in CD4+ T cells stimulated and allowed to 'rest' as in a, followed by activation overnight with plate-bound anti-CD3 and soluble anti-CD28 in the presence of a protein-transport inhibitor. Numbers in quadrants indicate percent cells in each (throughout). (e) Immunoblot analysis of T-bet and GATA-3 in lysates of CD4+ T cells stimulated for 48 h under TH1- or TH2-polarizing conditions (key) with plate-bound anti-CD3 and anti-CD28 and then allowed to 'rest' for 5 d in the presence of IL-2 (left), and intracellular staining of T-bet and GATA-3 in cells treated as at left and then fixed and permeabilized and assessed by flow cytometry (right). Numbers in plots (right) indicate mean fluorescence intensity. *P < 0.001 (analysis of variance (ANOVA)). Data are representative of four independent experiments (error bars (a–c), s.e.m. of triplicates).
Next we assessed the ability of T-Sgk1−/− T cells to produce IFN-γ and to be polarized into TH1 effector cells. Without any polarizing cytokines, T cells from C57BL/6 mice 'preferentially' differentiated into TH1 cells (Fig. 3d). Activation of wild-type and T-Sgk1−/− mice CD4+ T cells cultured under TH0 or TH1-polarizing conditions led to the generation of IFN-γ-producing cells (Fig. 3d). However, T-Sgk1−/− T cells had substantial IFN-γ expression even when they were differentiated under TH2-polarizing conditions (Fig. 3d). Thus, T cells that lacked SGK1 did not produce IL-4, IL-5 or IL-13 but instead inappropriately produced IFN-γ under TH2-polarizing conditions. Consistent with published studies12,13, loss of SGK1 did not affect TH17 polarization (Supplementary Fig. 4).
Next we investigated the lineage-specific transcription factors T-bet and GATA-3. As expected, wild-type T cells cultured under TH1-polarizing conditions had abundant expression of T-bet, while wild-type cells cultured under TH2-polarizing conditions had substantial expression of GATA-3 (Fig. 3e). In contrast, T-Sgk1−/− T cells had abundant expression of T-bet when cultured under either TH1-polarizing conditions or TH2-polarizing conditions. Such cells also failed to express GATA-3 under TH2-polarizing conditions. Overall, these observations demonstrated a role for SGK1 in reciprocally regulating TH1 differentiation and TH2 differentiation. During the differentiation of CD4+ effector T cells, SGK1 activation simultaneously enhanced IL-4 production and inhibited IFN-γ production.
SGK1 regulates TH2 differentiation by stabilizing JunB
We sought to define the biochemical mechanism by which SGK1 regulated TH1 and TH2 differentiation. We did not observe a defect in expression of the receptor for IL-4 or IL-4-induced phosphorylation of STAT6 in the absence of SGK1 (data not shown). Published studies of the role of SGK1 in renal epithelial cells have demonstrated that SGK1 negatively regulates the E3 ligase Nedd4-2 by phosphorylation at Ser342 and Ser448 (refs. 5,10). A closely related homolog of Nedd4-2 is the ubiquitin ligase Itch, which has been shown to interact with the adaptor Ndfip1 ('Nedd4 family–interacting protein 1') to mediate polyubiquitination of JunB26,27. JunB has been shown to be essential for the development of TH2 cells26,28. We sought to determine if the defect in TH2 differentiation that we observed in T-Sgk1−/− mice could have been due in part to increased ubiquitination and degradation of JunB by Nedd4-2. As shown before28, the abundance of JunB was higher in wild-type T cells cultured under TH2-polarizing conditions than in those cultured under TH1-polarizing conditions (Fig. 4a). In SGK1-deficient T cells, we observed that the abundance of JunB protein was decreased under both TH1-polarizing conditions and TH2-polarizing conditions (Fig. 4a). Those findings correlated with the phosphorylation status of Nedd4-2 at Ser342. That is, inhibition of Nedd4-2 (as measured by the phosphorylation of Nedd4-2 at Ser342) was greatest under TH2-polarizing conditions and was lower in the absence of SGK1 (Fig. 4a).

(a) Immunoblot analysis of JunB and of Nedd4-2 phosphorylated at Ser342 (p-Nedd4-2(S342)) (Nedd4-2 activity) in extracts of wild-type and T-Sgk1−/− CD4+ T cells stimulated in vitro under TH1- or TH2-polarizing conditions (left), and band density, normalized to loading controls and presented in arbitrary units (AU) relative to that of wild-type cells cultured under TH1-polarizing conditions, set as 1 (middle and right). (b) Intracellular staining of phosphorylated Nedd4-2 in OT-II (Thy-1.1+) CD4+ T cells adoptively transferred into wild-type (Thy-1.2+) recipient mice immunized with OVA-alum to induce a TH2 immune response, followed by removal of spleens on day 4 and stimulation of splenocytes with OVA peptide recognized by major histocompatibility complex class II (presented as mean fluorescence intensity (MFI)). (c) Immunoblot analysis of JunB in extracts of wild-type and T-Sgk1−/− cultured under TH2-polarizing conditions in the presence (+) or absence (−) of the proteasome inhibitor MG132 during the final 2 h of stimulation. (d) Immunoblot analysis (IB) of the ubiquitination (ub) of proteins immunoprecipitated (IP) with anti-JunB from wild-type and T-Sgk1−/− CD4+ T cells polarized with IL-4 and treated with MG132 (+) or not (−). (e) ELISA of IL-4 in supernatants of CD4+ T cells stimulated for 48 h under TH2-polarizing conditions, then transfected with nontargeting siRNA with scrambled sequence (Scr) or siRNA targeting Nedd4-2 or Ndfip (key), followed by population expansion for 5 d in IL-2 and restimulation overnight with anti-CD3 and anti-CD28. (f) Immunoblot analysis of Nedd4-2 and JunB in lysates of wild-type or T-Sgk1−/− CD4+ T cells treated as in e. *P = 0.0183 and **P < 0.001 (ANOVA). Data are representative of three (a–d), four (e) or two (f) independent experiments (error bars (a,e), s.e.m.).
We next investigated the ability of SGK1 to regulate Nedd4-2 in vivo. For this, we adoptively transferred congenically marked (Thy-1.1+) ovalbumin (OVA)-specific (OT-II) CD4+ T cells from wild-type or T-Sgk1−/− mice into naive hosts that we subsequently immunized with OVA adsorbed onto alum to induce a TH2 response. Similar to the results obtained in vitro, we observed phosphorylation of Nedd4-2 at Ser342 (and thus inhibition of Nedd4-2) after stimulation of wild-type adoptively transferred cells with the OVA epitope (amino acids 329–337) recognized by major histocompatibility complex class II (Fig. 4b). In contrast, Nedd4-2 was not phosphorylated nor inhibited in adoptively transferred T-Sgk1−/− cells.
Our data were consistent with the hypothesis that Nedd4-2 degraded JunB protein in T cells lacking SGK1 even under TH2-polarizing conditions, both in vitro and in vivo. To further test our hypothesis, we activated wild-type and T-Sgk1−/− CD4+ T cells under TH2-polarizing conditions in the presence of the proteasome inhibitor MG132. T cells lacking SGK1 had a much lower abundance of JunB than did wild-type T cells (Fig. 4c). However, the expression of JunB protein was restored in T-Sgk1−/− cells in the presence of MG132 (Fig. 4c), which suggested that JunB was expressed in T-Sgk1−/− T cells but was subsequently degraded. To confirm that the degradation of JunB was due to increased ubiquitination, we immunoprecipitated JunB from wild-type and T-Sgk1−/− CD4+ T cells, followed by immunoblot analysis of ubiquitin. We found that JunB was not ubiquitinated under TH2-polarizing conditions in wild-type cells, but ubiquitination of JunB was markedly enhanced in the absence of SGK1 (Fig. 4d).
Next we sought to determine whether we could restore the ability of T-Sgk1−/− CD4+ T cells to make IL-4 under TH2-polarizing conditions by knocking down Nedd4-2 or Ndfip through the use of small interfering RNA (siRNA). Knocking down either Nedd4-2 or Ndfip led to partial restoration of the production of IL-4 in TH2-polarized T-Sgk1−/− T cells (Fig. 4e). We confirmed the knockdown by measuring the expression of Nedd4-2 mRNA and Ndfip mRNA (Supplementary Fig. 5a,b). Additionally, we confirmed knockdown of Nedd4-2 at the protein level, which correlated with the reaccumulation of JunB protein (Fig. 4f). It was difficult to confirm knockdown of Ndfip at the protein level because the antibodies available lack sensitivity and specificity; however, knockdown of Ndfip also resulted in the reaccumulation of JunB protein (data not shown). Together these results suggested that activation of SGK1 promoted TH2 differentiation in part by negatively regulating E3 ligases that target JunB for degradation.
Resistance of T-Sgk1−/−mice to TH2 cell–mediated asthma
Since we had defined the role of SGK1 in regulating the differentiation of helper T cells in vitro, we wanted to confirm that paradigm in an in vivo model. We chose to study the OVA-alum model of allergic asthma because TH2 cells are involved in the early pathogenesis of this disease29 and we therefore hypothesized that T-Sgk1−/− mice would be resistant to allergic asthma. Wild-type mice mounted a stereotypical TH2 response to that allergic stimulus, characterized by IL-4 in bronchoalveolar lavage (BAL) fluid and immunoglobulin E (IgE) in serum (Supplementary Fig. 6a,b). However, BAL fluid and serum from T-Sgk1−/− mice contained significantly less IL-4 and IgE (Supplementary Fig. 6a,b). In contrast, we detected OVA-specific IgG2a, characteristic of a TH1 response, in the serum of T-Sgk1−/− mice (Supplementary Fig. 6c). Along those lines, lung lymphocytes from T-Sgk1−/− mice inappropriately produced IFN-γ when stimulated ex vivo (Supplementary Fig. 6d). Thus, in this in vivo model of TH2 cell–mediated inflammation, loss of SGK1 in T cells abrogated the TH2 response and instead resulted in an inappropriate TH1 response to an allergic stimulus.
Given those findings, we sought to determine if deletion of SGK1 in T cells would prevent allergen-induced lung pathology. For this, we challenged wild-type and T-Sgk1−/− mice with house dust mite (HDM) extract. Similar to our findings obtained with the OVA-alum adjuvant model, we observed a much lower TH2 response in T-Sgk1−/− mice than in wild-type mice after challenge with HDM extract (Fig. 5). Expression of IL-4, IL-13 and IL-5 in the lungs of T-Sgk1−/− mice was much lower than that in the lungs of wild-type mice (Fig. 5a–c). Likewise, we observed lower expression of those cytokines by CD4+ T cells in mediastinal lymph nodes from T-Sgk1−/− mice than in their wild-type counterparts (Fig. 5d–g). We observed a much lower absolute number of CD4+ T cells in lymph nodes from T-Sgk1−/− mice than in their wild-type counterparts; however, there was no significant difference between wild-type and T-Sgk1−/− mice in this parameter in the lungs or BAL fluid (Supplementary Fig. 6e,f). Consistent with our model, CD4+ T cells from the lungs, BAL fluid and lymph nodes of T-Sgk1−/− mice produced significantly more IFN-γ than did those from wild-type mice (Fig. 5h,i). Furthermore, the T-Sgk1−/− mice produced less total IgE, in addition to less HDM-specific IgE and IgG1, than did wild-type mice (Fig. 5j–l). Finally, the lungs of T-Sgk1−/− mice demonstrated much less goblet-cell hyperplasia and less peribronchiolar and perivascular inflammation than did the lungs of wild-type mice (Fig. 5m–q). Thus, loss of SGK1 in T cells protected against allergen-induced, TH2 cell–mediated lung disease.

(a–c) ELISA of IL-4 (a), IL-13 (b) and IL-5 (c) in supernatants of lung lymphocytes from wild-type and T-Sgk1−/− mice that had been sensitized intraperitoneally and rechallenged intranasally on days 14 and 21 with HDM extract (to induce allergic airway inflammation), followed by stimulation of lymphocytes for 3 d ex vivo with HDM extract. (d–g) Frequency of IL-4+ cells (d), IL-13+ cells (e), IL-5+ cells (f) or IL-5+IL-13+ cells (g) among lymphocytes obtained from mediastinal lymph nodes (LN) of mice immunized as in a–c and then stimulated for 4 h ex vivo with the phorbol ester PMA and ionomycin, followed by analysis of fixed and permeabilized cells by intracellular staining and flow cytometry. (h,i) Frequency of IFN-γ+ cells among lymphocytes obtained from the lung parenchyma or BAL fluid (h) or mediastinal lymph nodes (i) of wild-type and T-Sgk1−/− mice, then stimulated and assessed as in d–g. (j–l) Total IgE (j), HDM-specific IgE (k) and HDM-specific IgG1 (l) in serum from wild-type and T-Sgk1−/− mice sensitized and rechallenged with HDM extract as in a–c; antibody titers were extrapolated from a standard curve (if available) or results are presented as absorbance at 450 nm (A450) for a dilution of serum for which all samples were in the range of the assay. (m,n) Microscopy of lung sections obtained from wild-type and T-Sgk1−/− mice treated with saline control (Mock) or HDM and stained with periodic acid–Schiff reagent (m) or hematoxylin and eosin (n). Original magnification, ×10 (top and middle) or ×20 (bottom). (o,p) Histological scores of PAS+ cells (those that stained with periodic acid–Schiff reagent) from the large bronchioles (o) and small bronchioles (p) of HDM-treated mice. (q) Histological scores for perivascular and peribronchiolar inflammation in lung sections stained with hematoxylin and eosin as in n. Each symbol (a–l,o–q) represents an individual mouse; small horizontal lines indicate the mean (±s.e.m.). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 (Student's t-test). Data are representative of three (a–i), four (j–l) or two (m–q) independent experiments with five to eleven mice per group.
SGK1 negatively regulates IFN-γ production via TCF-1
Disruption of the SGK1–Nedd4-2–Ndfip–JunB axis suggests a possible mechanism for the failure of T-Sgk1−/− CD4+ T cells to differentiate toward a TH2 phenotype. However, it does not adequately explain the production of IFN-γ by CD4+ T cells under TH2 conditions in the absence of SGK1. It has been reported that the kinase GSK-3β is inhibited by the AGC kinases SGK1 and Akt9. In turn, GSK-3β phosphorylates β-catenin and targets it for degradation30,31. We observed much lower expression of β-catenin in T-Sgk1−/− T cells than in wild-type T cells (Fig. 6a). β-catenin is critical during TH2 differentiation because it promotes early GATA-3 expression by binding to the proximal promoter of the gene encoding this transcription factor32. In the absence of SGK1, GSK-3β remained highly active and thus there was very little total β-catenin (Fig. 6a). In contrast, inhibiting GSK-3β in T-Sgk1−/− T cells with LiCl led to an increase in total β-catenin relative to that obtained with no treatment (Fig. 6a).

(a) Immunoblot analysis of β-catenin phosphorylated at Ser33, Ser47 and Thr41 (p-β-catenin) and total β-catenin, as well as total GSK-3β (loading control), in extracts of wild-type and T-Sgk1−/− CD4+ T cells left unstimulated (IL-4 −) or stimulated under TH2-polarizing conditions (IL-4 +) and left untreated (LiCl −) or treated overnight (LiCl +) with 25 mM LiCl. (b) Immunoblot analysis of the long and short isoforms (right margin) of TCF-1 in extracts of wild-type and T-Sgk1−/− CD4+ T cells stimulated in vitro under TH1- or TH2-polarizing conditions, presented as short and long exposures of the blots (left margin). (c) Quantitative PCR analysis of the long isoform of TCF-1 in CD4+ T cells purified from wild-type and T-Sgk1−/− mice by magnetic separation, then stimulated for 2 d with anti-CD3 and anti-CD28 magnetic beads in TH1- or TH2-polarizing conditions (key) and assessed at 72 h or 8 d after stimulation; results were normalized those of 18S rRNA and are presented relative to those of wild-type cells cultured under TH1-polarizing conditions for 72 h, set as 1. (d) Flow cytometry of CD4+ T cells cultured under TH2-polarizing conditions and transduced with empty vector (EV) or vector encoding the long isoform of TCF-1 and a human CD8 reporter (FL–TCF-1), then allowed to 'rest' and then sorted for surface expression of human CD8 (middle), followed by restimulation of sorted cells and intracellular staining of IFN-γ (top and bottom). Outlined areas (middle) indicate gating on low (left), intermediate (middle) and high (right) expression of human CD8 (FSC, forward scatter). Numbers above bracketed lines (bottom) indicate percent IFN-γ+ cells from T-Sgk1−/− mice (among cells gated on high expression of human CD8). * <P < 0.001 (ANOVA). Data are representative of three (a–c) or four (d) independent experiments (error bars (c), s.e.m. of triplicates).
The transcription factor TCF-1 is both a downstream target of β-catenin and a transcriptional coactivator with β-catenin31,32,33. Similar to T-Sgk1−/− mice, mice deficient in TCF-1 produce more IFN-γ than do wild-type mice, even under TH2-polarizing conditions32. Specifically, the long isoforms of TCF-1 contain an additional β-catenin-binding domain, and those long isoforms promote TH2 polarization by increasing GATA-3 expression and repressing IFN-γ expression32. We hypothesized that T cells lacking SGK1 would have lower expression of the long form of TCF-1 than would their wild-type counterparts. We cultured wild-type and T-Sgk1−/− CD4+ T cells under TH1- or TH2-polarizing conditions and analyzed their TCF-1 protein. We found that T-Sgk1−/− cells had a lower abundance of the long isoforms of TCF-1 than did their wild-type counterparts under each condition, but there was no difference between T-Sgk1−/− cells and wild-type cells in their expression of the short isoforms of TCF-1 (Fig. 6b).
To determine if SGK1 regulates TCF-1 expression at the transcriptional level, we designed primers that flank the β-catenin-binding domain to specifically detect the long isoform of TCF-1. We observed less transcription of the long isoforms of TCF-1 at 72 h after activation of the T cell antigen receptor in SGK1-deficient cells than in wild-type cells and observed a complete absence of transcripts at 8 d after such stimulation (Fig. 6c). To further test our hypothesis, we sought to 'rescue' the phenotype of T-Sgk1−/− CD4+ T cells by retroviral transduction with a vector encoding the long isoform of TCF-1 and a human monomorphic coreceptor CD8 reporter. We sorted transduced cells for expression of human CD8, then restimulated the cells with anti-CD3 and anti-CD28 and measured the production of IFN-γ by intracellular staining. Wild-type T cells cultured under TH2-polarizing conditions did not express IFN-γ (Fig. 6d). T-Sgk1−/− T cells transduced with the empty vector had substantial expression of IFN-γ under TH2-polarizing conditions (Fig. 6d). In contrast, IFN-γ expression was diminished in T-Sgk1−/− T cells transfected with vector encoding the long isoform of TCF-1 and the reporter (Fig. 6d). These data supported a model whereby under TH2-polarizing conditions, SGK1 inhibited the GSK-3β-mediated degradation of β-catenin, which led in turn to higher expression of the long form of TCF-1 and inhibition of IFN-γ (Supplementary Fig. 7).
T-Sgk1−/− mice mount robust TH1 cell–mediated immune responses
The ability of SGK1 to regulate TH2 responses is due in part to its ability to negatively regulate IFN-γ. We sought to determine whether T-Sgk1−/− mice would also produce more IFN-γ in vivo under strongly TH1-polarizing conditions. For this, we adoptively transferred congenically marked Thy-1.1+, OVA-specific wild-type and T-Sgk1−/− CD4+ T cells into Thy-1.2+ wild-type mice that we simultaneously infected intravenously with OVA-expressing vaccinia virus, which induces robust TH1 immune responses. Infection with that virus led to a greater abundance of OVA-specific, IFN-γ-producing cells in T-Sgk1−/− mice than in wild-type mice (Fig. 7a). In addition, the TH1 response in T-Sgk1−/− mice was characterized by a greater amount of IFN-γ produced (on a per-cell basis) than that produced by their wild-type counterparts (Fig. 7a). To confirm those observations, we challenged wild-type and T-Sgk1−/− mice with the PR8 strain of influenza virus, which also promotes a strong TH1-mediated immune response. Similar to the results we obtained with vaccinia virus, in response to infection with influenza virus, CD4+ T cells from T-Sgk1−/− mice produced more IFN-γ than did their wild-type counterparts (Fig. 7b).

(a) IFN-γ production by wild-type and T-Sgk1−/− OT-II cells adoptively transferred into wild-type host mice (n = 10 per group) simultaneously immunized intravenously with 1 × 106 plaque-forming units of OVA-expressing vaccinia virus, followed by restimulation of the cells ex vivo with PMA and ionomycin; gates for IFN-γ were set with a 'no-stimulation' control such that <1.0% of cells were allowed in the positive gate. Numbers above bracketed lines (left) indicate percent IFN-γ+ cells; numbers in top right corners indicate mean fluorescence intensity of IFN-γ. Right, quantification of the results at left. (b) IFN-γ production by lung lymphocytes obtained from wild-type and T-Sgk1−/− mice 8 d after intranasal infection with the PR8 strain of influenza virus, followed by stimulation of the cells for 4 h ex vivo with PMA and ionomycin, then analysis of fixed and permeabilized cells by intracellular staining and flow cytometry. Each symbol (right) represents an individual mouse; small horizontal lines indicate the mean (±s.e.m.). (c) Quantification of B16 metastases in lungs of wild-type and T-Sgk1−/− mice 21 d after intravenous injection of 2 × 105 B16 melanoma cells. (d) Production of IFN-γ by CD4+ T cells among lung lymphocytes as in b. (e) Survival of wild-type and T-Sgk1−/− mice (n = 5–7 per group) vaccinated with 1 × 106 plaque-forming units of OVA-expressing vaccinia virus 7 d before challenge with 2 × 105 OVA-expressing B16 melanoma cells, assessed by Kaplan-Meier analysis. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 (Student's t-test (a–d) or log-rank (Mantel-Cox) analysis (e)). Data are representative of two to three independent experiments with five to fifteen mice per group (error bars (a,c,d), s.e.m.).
To further assess the TH1 response of T-Sgk1−/− cells in vivo, we used a model of TH1 cell–mediated tumor rejection, since TH1-polarized CD4+ T cells have been shown to serve an important role in antitumor immunity34. To address that, we injected 2 × 105 B16 mouse melanoma cells intravenously into wild-type and T-Sgk1−/− mice and collected their lungs 21 d later. T-Sgk1−/− mice had less than half as many lung tumors as wild-type mice had (Fig. 7c and Supplementary Fig. 8). While it is possible that the lower tumor burden could have been promoted by CD8+ T cells, it was apparent that the more effective response of the T-Sgk1−/− mice was associated with a greater abundance of IFN-γ-producing CD4+ T cells in the lungs (Fig. 7d). Next we sought to further boost tumor immunity in T-Sgk1−/− mice by vaccination. We immunized mice with OVA-expressing vaccinia virus 7 d before intravenously injecting OVA-expressing B16 cells, then monitored survival as the main outcome. We found that T-Sgk1−/− mice survived significantly longer than did wild-type mice in response to the tumor vaccine (Fig. 7e). Thus, targeted deletion of SGK1 in T cells led to enhanced tumor immunity characterized by more production of IFN-γ and robust immunological rejection of tumors.
Discussion
A critical task of the adaptive and innate immune systems is to integrate cues from the immunological microenvironment and then respond appropriately. It has become clear that the evolutionarily conserved serine-threonine kinase mTOR has a central role in this process15. The outcome of the activation of mTOR is determined by the regulation of selective downstream signaling pathways and substrates. We have identified the AGC kinase SGK1 as an mTORC2-activated substrate that selectively regulated the differentiation of TH1 and TH2 cells. T cells lacking SGK1 demonstrated a markedly diminished ability to differentiate into TH2 cells both in vitro and in vivo. Furthermore, under TH2-polarizing conditions, T-Sgk1−/− T cells inappropriately produced IFN-γ. Thus, under TH2-polarizing conditions, SGK1 simultaneously promoted TH2 differentiation while also inhibiting the production of TH1 cytokines. Likewise, we observed greater IFN-γ production by T-Sgk1−/− T cells in vivo in the setting of viral infection and antitumor immunity. Of note, under TH1-polarizing conditions in vitro, T-Sgk1−/− and wild-type T cells produced equivalent amounts of IFN-γ. Perhaps that indicated that under strongly polarizing conditions in vitro, the ability of SGK1 to inhibit the TH1 response was inconsequential. However, under in vivo inflammatory conditions that predominantly favored TH1 polarization, signaling by SGK1 seemed to limit the magnitude of the TH1 immune response.
Our studies have defined a mechanism by which activation of mTOR (specifically mTORC2) regulated TH1 and TH2 differentiation. It has been reported that SGK1 has a role in promoting the stability of pathogenic TH17 cells12. Notably, in that study, as in ours, TH17 differentiation was similar in both wild-type and T-Sgk1−/− T cells. However, that study demonstrated a role for SGK1 in controlling expression of the IL-23 receptor12. As a result, T cells lacking SGK1 respond poorly to IL-23, and T-Sgk1−/− mice are resistant to the development of experimental autoimmune encephalitis12. Furthermore, an increase in the extracellular concentration of sodium leads to increased IL-23-mediated TH17 differentiation and that effect is abrogated in T-Sgk1−/− T cells12. The authors of that report12 and another study13 attribute their findings to the ability of an increase in dietary salt to exacerbate experimental autoimmune encephalitis.
Published studies of the role of SGK1 in the kidney have provided clues about how this AGC kinase may function in T cells11. To allow sodium to be reclaimed in the urine, SGK1 prevents the degradation of sodium channels by phosphorylating and inhibiting the E3 ubiquitin ligase Nedd4-2 (refs. 5,6). Similarly, we found that SGK1 promoted TH2 differentiation by phosphorylating and inhibiting Nedd4-2, thus preventing degradation of JunB. Those findings are consistent with studies reporting that mice that lack Ndfip or Itch display a 'hyper-TH2' phenotype26,27. In those studies, loss of Ndfip or Itch led to the accumulation of JunB and increased expression of IL-4. Overall, our studies serve to strengthen the link between mTOR-SGK1 and the regulation of cellular differentiation and function by E3 ligases. Just as the ability of mTOR to regulate E3 ligase activity has been shown to be important in T cells and renal epithelial cells, we propose that these regulatory pathways will prove to be important for diverse cellular functions.
Our findings have identified SGK1 as a potential target for the treatment of TH2 cell–mediated autoimmune and allergic diseases, such as asthma. At present, nonspecific immunosuppressive agents such as steroids serve an important role in the treatment of acute asthma flares, as well as in the prevention of recurrences35,36,37. Our data suggest that selective inhibition of SGK1 might represent an immunomodulatory approach for treating and preventing the sequelae of asthma and other allergic diseases without globally inhibiting the immune system. Notably, small-molecule inhibitors of SGK1 have already been developed38,39. Additionally, inhibiting SGK1 may be of therapeutic value in terms of enhancing TH1 cell–mediated immune responses, such as in the setting of viral infection or as an adjunct to tumor immunotherapy. Thus, whereas an asthmatic patient might be treated with an inhibitor of SGK1 to prevent the development of TH2 cell–mediated inflammation, the same small-molecule inhibitor might be given to a patient during cancer immunotherapy to enhance the antitumor response.
Methods
Mice.
Mice were kept in accordance with guidelines of the Johns Hopkins University Institutional Animal Care and Use Committee. Mice with loxP-flanked Sgk1 alleles have been described11, and mice with loxP-flanked Rictor alleles were provided by M. Magnuson40. C57BL/6 mice, mice expressing Cre recombinase from the Cd4 promoter and OT-II mice from Jackson Laboratories were bred to Thy-1.1+ backgrounds. 5C.C7 mice were from Taconic Farms.
Antibodies and reagents.
The following antibodies for flow cytometry were from BD Biosciences: anti-B220 (RA3-6B2), anti-CD3 (2C11), anti-CD4 (RM4-5), anti-CD8 (Ly-3), anti-CD25 (PC61), anti-CD44 (IM7), anti-CD124 (mIL4R-M1), anti-IFN-γ (XMG1.2), anti-IL-17a (TC11-18H10) and anti-human CD8 (RPA-T8). Anti-CD62L (MEL-14), anti-IL-13 (eBio13A), anti-IL-5 (TRFK5), anti-T-bet (eBio4B10) and anti-Foxp3 (FJK-16s) were from eBioscience.
The following antibodies for immunoblot analysis were from Cell Signaling Technologies: anti-rictor (2140), antibody to NDRG1 phosphorylated at Thr346 (5482), anti-NDRG1 (9408), antibody to Akt phosphorylated at Ser473 (4060), anti-Akt (4685), antibody to Nedd4-2 phosphorylated at Ser342 (4080), anti-Nedd4-2 (4013), anti-TCF-1 (2206), anti-β-catenin (9562), antibody to phosphorylated β-catenin (9561) and antibody to phosphorylated STAT6 (9361). Anti-T-bet (4B10) and anti-GATA-3 (TWAJ) were from eBioscience. Anti-JunB (210) was from Santa Cruz Biotechnology. Anti-ubiquitin (U5379) and anti-actin (A2066) were from Sigma. MG132 was from Sigma, PP242 was from Calbiochem and CFSE (carboxyfluorescein diacetate succinimidyl ester) was from Invitrogen.
T cell stimulation and polarization.
CD4+ T cells were purified by negative selection with a CD4+ isolation kit and MACS cell separation (Miltenyi Biotec). For polarizing experiments, T cells were stimulated with irradiated autologous antigen-presenting cells (APCs) or with plate-bound anti-CD3 and soluble anti-CD28. For APC-free stimulations, flat-bottomed plates were coated with anti-CD3 (3 μg/ml) diluted in PBS, and soluble anti-CD28 (2 μg/ml) was added to the cultures. For stimulation with APCs, T cells were cultured with irradiated APCs and anti-CD3 (1 μg/ml), followed by rest and population expansion for 5 d. For rechallenge, live cells were harvested through the use of Ficoll gradients and washed, then were stimulated with plate-bound anti-CD3 (1 μg/ml) and soluble anti-CD28 (2 μg/ml). Polarizing conditions were as follows: TH0, anti-IL-4 (5 μg/ml) and anti-IFN-γ (5 μg/ml), with only IL-2 (1 ng/ml) during the rest period; TH1, IL-12 (5 ng/ml), IFN-γ (100 ng/ml) and anti-IL-4 (5 μg/ml), with only IL-2 (1 ng/ml) during the rest period; TH2, IL-4 (10 ng/ml), anti-IL-12 (5 μg/ml) and anti-IFN-γ (5 μg/ml), with only IL-2 (1 ng/ml) during the rest period; TH17, IL-6 (30 ng/ml), TGF-β (3 ng/ml), anti-IFN-γ (5 μg/ml), anti-IL-4 (5 μg/ml), with only IL-23 (10 ng/ml) during the rest period. Stimulatory anti-CD3 (2C11) and anti-CD28 (37.51), as well as neutralizing anti-IFN-γ (XMG1.2) and anti-IL-4 (11B11), were purified from hybridoma supernatants prepared in-house. Neutralizing anti-IL-12p40 (C17.8) was from eBioscience. Cytokines were from Peprotech.
Adoptive transfer.
1 × 106 naive CD4+ T cells specific for the OVA epitope (amino acids 329-337) recognized by major histocompatibility complex class II and congenically marked with Thy-1.1 were transferred intravenously into Thy-1.2+ C57BL/6 recipients by injection into theretro-orbital sinus. OVA-expressing vaccinia virus (1 × 106 plaque-forming units) was simultaneously administered by either injection in to the retro-orbital sinus or intraperitoneal injection.
HDM asthma model.
On days 0 and 7, mice were primed by intraperitoneal injection of 10 μg HDM extract (Dermatophagoides pteronyssinus; Greer Labs) in PBS. On days 14 and 21, mice were challenged by inhalation of HDM extract (50 μg per nostril). On day 24, mice were killed for analysis. Lymphocytes from BAL fluid and mediastinal lymph nodes were stimulated for 4 h ex vivo with PMA (phorbol 12-myristate 13-acetate) and ionomycin. Lung lymphocytes were stimulated for 72 h ex vivo with 30 μg HDM extract, and supernatants of cultures were analyzed by ELISA. Differential cellular analysis of BAL fluid was achieved by cytospin and staining with Diff-Quick. For histological analysis, lungs were fixed with formalin, embedded in paraffin and stained with hematoxylin and eosin, or periodic acid–Schiff reagent and were assigned scores by published methods41. Peribronchiolar and perivascular inflammation was determined by a semiquantitatively graded scale as follows: 0, no inflammation; 1, mild inflammation; 2, moderate inflammation; 3, severe. Goblet-cell hyperplasia was assessed by staining of histologic sections with periodic acid–Schiff reagent (PAS) and was semiquantitatively graded as follows: 0, no or rare PAS+ cells; 1, <10% PAS+ cells observed at a magnification of ×10; 2, 10–25% PAS+ cells observed at a magnification of ×10X and 3, >25% PAS+ cells observed at a magnification of ×4. A composite score was determined by multiplication of the graded score and the frequency of airways or vessels with a given pathological finding. All scores were assigned by a pathologist (P.I.) 'blinded' to the identity of the samples, and all slides were examined in a random order.
Influenza model.
On day 0, mice were challenged with intranasal injection of influenza strain PR8 (Charles River) at a concentration of 600 egg-infective dose (EID) per nostril for a final dose of 1,200 egg-infective dose. On day 8, mice were killed. Lymphocytes from the lungs were collected by gentle mechanical and chemical digestion of lung parenchyma with collagenase and DNAse and were stimulated for 4 h ex vivo with PMA and ionomycin. Experimental samples from groups were excluded from the final analysis if analysis of T lymphocytes from lungs by flow cytometry demonstrated more than 70% CD44− cells and no detectable cells positive for a tetramer specific for the nucleoprotein antigen of influenza virus, suggestive of inadequate inoculation with influenza virus.
B16 melanoma model.
2 × 105 B16-OVA cells were injected intravenously in mice on day 0 and lungs were collected 21 d later. Lung lymphocytes were stimulated ex vivo with PMA and ionomycin (Sigma) at a concentration of 50 ng/ml and 500 ng/ml, respectively. For survival experiments, 1 × 106 plaque-forming units of VAC-OVA were injected intraperitoneally into mice on day 0, followed by intravenous injection of 2 × 105 B16-OVA cells on day 7.
OVA airway-sensitization model.
OVA (Sigma-Aldrich) was adsorbed onto Imject Alum adjuvant (Pierce) by gentle shaking for 30 min, to a final concentration of 250 μg/ml. Mice were primed by intraperitoneal injection of 50 μg of OVA-alum in 200 μl. On day 7, mice were 'boosted' by the same protocol. On days 15, 16, and 17, mice were challenged by intranasal injection of 25 μg OVA in PBS per nostril. On day 18, mice were killed.
Intracellular staining.
Brefeldin A (GolgiPlug; BD Biosciences) or monensin (GolgiStop; BD Biosciences) was used for cytokine staining. Surface antigens on cells were stained, then were fixed and made permeable with either a Cytofix/Cytoperm kit (BD) or a Fixation/Permeabilization kit (eBioscience), then intracellular cytokines in cells were stained. For intracellular staining of phosphorylated proteins, cells were fixed with 2.0% formalin and permeabilized with methanol.
Flow cytometry.
Flow cytometry data were acquired with a FACSCalibur or LSR II (BD) and were analyzed with FlowJo7.6 software (TreeStar). Gates were determined through the use of unstimulated control cells or isotype-matched control antibodies where appropriate.
Cells were sorted with a FACSAria (BD) at the Flow Cytometry Core at the Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine.
Immunoblot analysis.
For immunoblot analysis of mTOR substrates, CD4+ T cells were stimulated in 'RPMI medium–based T cell' medium with anti-CD3 (1 μg/ml), anti-CD28 (2 μg/ml) and antibody to hamster immunoglobulin IgG1 (0.75 μg/ml; G94-56; BD Biosciences). T cells were harvested by centrifugation and were resuspended in ice-cold lysis buffer (20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate (glycerol-2-phosphate), 1 mM sodium orthovanadate, 1 mM PMSF and 1× protease inhibitors (Roche)) and were lysed for 30 min at 4 °C. Lysates were cleared of debris by high-speed centrifugation. An equal protein mass from each condition was mixed with 4× LDS buffer (Invitrogen) and samples were boiled for 10 min. Lysates were then loaded into NuPAGE gels (10% Bis–Tris gels; Invitrogen) and were separated by electrophoresis at 150 V for 90 min. Proteins were transferred for 90 min at 30 V onto polyvinylidene fluoride membranes with transfer buffer (1× NuPAGE Transfer Buffer (Invitrogen) with 20% methanol). Nonspecific binding was blocked by incubation of membranes for 60 min in 5% nonfat dry milk, then membranes were washed briefly with 0.1% Tween-20 in Tris-buffered saline, pH 8 (TBST), and were probed overnight at 4 °C with primary antibody in 4% bovine serum albumin in TBST. Membranes were washed three times, 10 min each, with TBST and were probed with horseradish peroxidase–conjugated secondary antibody in nonfat dry milk. Membranes were washed two times in TBST, 5 min each, and then once for 5 min in Tris-buffered saline. ECL plus substrate (GE Healthcare) was used for detection of horseradish peroxidase–labeled antibodies. Blots were developed with the Biospectrum Multispectrum Imaging System, and images were acquired and analyzed with ImageJ software (NIH) or VisionWorks, LS Image Acquisition and Analysis software (UVP).
ELISA.
IL-2, IL-4, IL-5 and IL-13 were analyzed by ELISA as recommended by the manufacturer (eBioscience). Total serum IgE was measured with the following reagents from BD: anti–mouse IgE (capture antibody; 553413), mouse IgE standard (557080) and biotinylated anti-mouse IgE (detection antibody; 553419). HDM and OVA-specific antibodies were analyzed by ELISA in Nunc-Immuno plates that had been coated overnight at 4 °C with 100 μl HDM (25 μg/ml in PBS) or OVA (20 μg/ml in PBS), with nonspecific binding blocked by incubation for 1 h at 25 °C with assay diluent (eBioscience). Wells were washed several times and mouse serum samples diluted in assay diluent were added for 2 h or overnight. Wells were washed, then biotinylated antibody to mouse immunoglobulin IgG1a (553441; BD) or to IgG2a (553455; BD), diluted to a concentration of 0.5 μg/ml in assay diluent, was added for 1 h. Wells were washed, then streptavidin-conjugated horseradish peroxidase (eBioscience) was added for 30 min. Wells were washed seven times, then were incubated for 15 min with tetramethylbenzidine substrate or until the reaction approached saturation. Linear-regression analysis or measurement of absorbance was used to determine antibody titers or relative differences.
Real-time PCR.
RNA was isolated and then was reverse-transcribed into cDNA with the SuperScript III First-Strand Synthesis System for RT-PCR according to the manufacturer's protocol (Invitrogen). Primers for short and long isoforms of TCF-1 were from Sigma, and primers to detect Nedd4-2 and Ndfip were from Applied Biosystems. Primers used to detect TCF-1 long isoforms were as follows: 5′-CAATCTGCTCATGCCCTACC-3′ (forward) and 5′-GCCTGTGAACTCCTTGCTTC-3′ (reverse). Long and short isoforms were quantified by real-time PCR with a SYBR green fluorescent probe (Applied Biosystems). A Step One Plus Real Time PCR System (Applied Biosystems) was used for quantitative RT-PCR.
Retroviral transduction.
T cells were stimulated overnight with plate-bound anti-CD3 and soluble anti-CD28 (as described above) before 'spin-infection' with supernatants from 293T Phoenix cells. The Phoenix helper-free retroviral ecotropic packaging cell line (Nolan Laboratory) was transfected with plasmid encoding MSCV retrovirus containing sequence encoding full-length TCF-1 and a human CD8 reporter (MSCV constructs provided by J.M.-S.), through the use of Lipofectamine 2000 (Invitrogen).
siRNA-mediated knockdown.
T cells were stimulated for 48 h with plate-bound anti-CD3 and soluble anti-CD28 (as described above) before transfection. For RNA-mediated interference, ON-TARGET siRNA reagents for control siRNA with a scrambled sequence and Nedd4-2- and Ndfip-specific siRNA were from Dharmacon. Nucleofection reagents were from Lonza.
Statistical analysis.
Prism software version 5.0 (GraphPad Software) was used for statistical analyses, including unpaired Student's t-test, two-way analysis of variance (ANOVA) and log-rank (Mantel-Cox) analysis. A P value of <0.05 was considered statistically significant.
References
Pearce, L.R., Komander, D. & Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell. Biol. 11, 9–22 (2010).
Biondi, R.M., Kieloch, A., Currie, R.A., Deak, M. & Alessi, D.R. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 20, 4380–4390 (2001).
Garcia-Martinez, J.M. & Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 416, 375–385 (2008).
Yan, L., Mieulet, V. & Lamb, R.F. mTORC2 is the hydrophobic motif kinase for SGK1. Biochem. J. 416, e19–e21 (2008).
Debonneville, C. et al. Phosphorylation of Nedd4–2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 20, 7052–7059 (2001).
Ichimura, T. et al. 14–3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4–2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 (2005).
Di Pietro, N. et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) regulates adipocyte differentiation via forkhead box O1. Mol. Endocrinol. 24, 370–380 (2010).
Brunet, A. et al. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 21, 952–965 (2001).
Sakoda, H. et al. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J. Biol. Chem. 278, 25802–25807 (2003).
Wiemuth, D. et al. Interaction of serum- and glucocorticoid regulated kinase 1 (SGK1) with the WW-domains of Nedd4–2 is required for epithelial sodium channel regulation. PLoS One 5, e12163 (2010).
Fejes-Toth, G., Frindt, G., Naray-Fejes-Toth, A. & Palmer, L.G. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am. J. Physiol. Renal Physiol. 294, F1298–F1305 (2008).
Wu, C. et al. Induction of pathogenic T17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517 (2013).
Kleinewietfeld, M. et al. Sodium chloride drives autoimmune disease by the induction of pathogenic T17 cells. Nature 496, 518–522 (2013).
Zoncu, R., Efeyan, A. & Sabatini, D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell. Biol. 12, 21–35 (2011).
Powell, J.D., Pollizzi, K.N., Heikamp, E.B. & Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 30, 39–68 (2012).
Delgoffe, G.M. et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009).
Zhang, S. et al. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood 117, 1228–1238 (2011).
Delgoffe, G.M. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303 (2011).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Murray, J.T. et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488 (2004).
Lu, M., Wang, J., Ives, H.E. & Pearce, D. mSIN1 protein mediates SGK1 protein interaction with mTORC2 protein complex and is required for selective activation of the epithelial sodium channel. J. Biol. Chem. 286, 30647–30654 (2011).
Feldman, M.E. et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 7, e38 (2009).
Sarbassov, D.D., Guertin, D.A., Ali, S.M. & Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
Guertin, D.A. et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871 (2006).
Jacinto, E. et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 (2006).
Oliver, P.M. et al. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity 25, 929–940 (2006).
Fang, D. et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat. Immunol. 3, 281–287 (2002).
Li, B., Tournier, C., Davis, R.J. & Flavell, R.A. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 18, 420–432 (1999).
Huang, T.J. et al. Allergen-specific Th1 cells counteract efferent Th2 cell-dependent bronchial hyperresponsiveness and eosinophilic inflammation partly via IFN-γ. J. Immunol. 166, 207–217 (2001).
Liu, C. et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 (2002).
Yu, Q., Sharma, A. & Sen, J.M. TCF1 and β-catenin regulate T cell development and function. Immunol. Res. 47, 45–55 (2010).
Yu, Q. et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-γ. Nat. Immunol. 10, 992–999 (2009).
Roose, J. et al. Synergy between tumor suppressor APC and the β-catenin-Tcf4 target Tcf1. Science 285, 1923–1926 (1999).
Quezada, S.A. et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 207, 637–650 (2010).
Suissa, S., Ernst, P., Benayoun, S., Baltzan, M. & Cai, B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N. Engl. J. Med. 343, 332–336 (2000).
Fanta, C.H. Asthma. N. Engl. J. Med. 360, 1002–1014 (2009).
Littenberg, B. & Gluck, E.H. A controlled trial of methylprednisolone in the emergency treatment of acute asthma. N. Engl. J. Med. 314, 150–152 (1986).
Sherk, A.B. et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 68, 7475–7483 (2008).
Ackermann, T.F. et al. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol. Biochem. 28, 137–146 (2011).
Kumar, A. et al. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol. Cell Biol. 28, 61–70 (2008).
Daan de Boer, J. et al. Lipopolysaccharide inhibits Th2 lung inflammation induced by house dust mite allergens in mice. Am. J. Respir. Cell. Mol. Biol. 48, 382–389 (2013).
Acknowledgements
We thank D. Pardoll, C. Gamper and members of the Powell Lab for discussions, and the Magnuson laboratory for Rictor−/− mice. Supported by the US National Institutes of Health (R01 AI77610 to J.D.P., NHLBI P01HL010342 and NHLBI R21HL111783 to M.R.H., DK 41481 to A.N.-F.-T. and DK 58898 to G.F.-T.), the American Asthma Foundation (J.D.P.), the Intramural Research Program of the NIH, National Institute on Aging (J.M.S.), the FAMRI Center of Excellence 108595 (M.R.H.) and the American Medical Association (E.B.H.).
Author information
Authors and Affiliations
Contributions
E.B.H. helped design and do experiments and write the paper; C.H.P., S.C., M.-H.O. and I.-H.S. assisted in the in vivo experiments and biochemistry; A.W. contributed to the design of experiments and interpretation of data; P.I. assigned scores to the lung histology; A.S. and J.M.-S. constructed the plasmid encoding TCF1 and helped with analysis of TCF1; A.N.-F.-T. and G.F.-T. generated the Sgk1fl/fl mice; M.R.H. helped design the lung experiments and contributed to the writing of the paper; and J.D.P. conceived of the project and helped design the experiments and write the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Sgk1 mRNA is expressed in CD4+ and CD8+ T cells and is upregulated in response to cytokines.
(a) CD4+ and CD8+ T cells were isolated from wild-type and T-Sgk1-/- mice by magnetic separation, and RNA was isolated. Sgk1 mRNA levels were determined by polymerase chain reaction using the following excision primers: forward primer 5-CTCAGTCTCTTTTGGGCTCTTT-3 and the reverse primer 5-TTTCTTCTTCAGGATGGCTTTC-3, as described in the Methods. GAPDH is included as a loading control. Data is representative of 3 independent experiments. (b) CD4+ T cells were isolated from 5C.C7 mice using magnetic separation, and cells were stimulated with anti-CD3 and anti-CD28 for 1 hour under polarizing conditions. RNA was isolated and reverse transcribed into cDNA to measure expression of Sgk1 by RT-PCR. Expression is normalized to the unstimulated control and to 18s rRNA expression
Supplementary Figure 2 mTORC2 is required for activation of SGK1.
(a) Model depicting downstream targets of mTORC2. Activation of mTORC2 leads to phosphorylation of Akt (p-S473) and SGK1 (p-S422). Activation of SGK1 leads to phosphorylation of its downstream target NDRG1 (p-T346). (b) ImageJ software was used to calculate band density from 3 independent experiments shown in Fig. 2a, and band density was normalized to loading controls and to WT unstimulated (US) conditions. (c) Flow cytometric analysis of CD4+ T cells isolated from WT, T-Rictor-/-, and T-Sgk1-/- mice and stimulated with anti-CD3 and anti-CD28 for 1,3, or 6 hours. Cells were permeabilised with methanol and interrogated for phosphorylation of Akt (S473) using flow cytometry. Numbers in upper right corner of each plot indicate mean fluorescent index (MFI) of p-Akt or total Akt. Below, MFI of p-Akt from flow cytometry plots above were normalized to the MFI of total Akt. Data are representative of 3 independent experiments (a-c), statistical significance calculated by ANOVA, ns=no significance, error bars s.e.m.)
Supplementary Figure 3 Characterization of lymphocyte development and maturation of T-Sgk1-/- mice.
(a) Flow cytometric phenotyping data compiled from multiple mice (n=6) of double negative (CD4-CD8-), double positive (CD4+CD8+), or single positive (CD4+ or CD8+) thymocytes, (b,c) absolute numbers of B220+, CD3+, CD4+, and CD8+ cells in the spleen and lymph nodes, (d-g) expression of CD44 and CD62L on both CD4+ and CD8+ subsets in the spleen and lymph nodes, (h) expression of Treg phenotypic markers (CD3+ CD4+ CD25+ FoxP3+) in spleen and lymph nodes. Data are compiled from 6 mice and are representative of 3 independent experiments. Statistical significance determined by Student's t-test, NS=No Significance, *P<0.05 **P<0.01, error bars s.e.m.
Supplementary Figure 4 SGK1 is not required for TH17 differentiation in vitro.
Intracellular staining of CD4+ T cells that were polarized for 48 hours under TH0 or TH17 (IL-6 and TGF-β) conditions, then rested and restimulated as in Fig. 3D. Data is representative of 3 independent experiments.
Supplementary Figure 5 Knockdown of Nedd4-2 and Ndfip by siRNA.
Quantitative PCR analysis of NEDD4-2 (a) and Ndfip (b) mRNA in CD4+ T cells from WT and T-Sgk1-/- mice. RNA was harvested from cells 24 hours after transfection with siRNA for analysis by real time PCR. Gene expression is normalized to 18s rRNA and to the WT scrambled control. Data is representative of two independent experiments, and samples were analyzed in triplicate for each experiment. Statistical significance calculated by ANOVA, *P<0.001, error bars s.e.m.)
Supplementary Figure 6 Diminished TH2 cell–mediated immunity in vivo in T-Sgk1-/- mice in asthma models.
(a) IL-4 production in bronchoalveolar lavage (BAL) harvested from mice that had been sensitized with OVA in alum i.p., then rechallenged with intranasal OVA on days 15, 16, and 17, followed by sacrifice on day 18. Lungs were lavaged with PBS and analyzed by ELISA. (b) Total IgE in serum. (c) OVA-specific IgG2a in serum. (d) IFN-γ production by lung lymphocytes, as measured by intracellular staining and flow cytometry. As in A, lung lymphocytes were harvested from diseased mice and stimulated for 4 hours ex vivo with PMA and ionomycin. (e) Analysis of CD4+ lymphocytes found in the lungs and mediastinal lymph nodes of mice that were sensitized and challenged with HDM extract as described in Fig 5. Graph depicts absolute number of CD4+ T cells recovered from each compartment. (f) BAL was analyzed by cytospin and Diff-quick staining for the presence of lymphocytes in WT and T-Sgk1-/- mice that were sensitized and challenged with HDM extract as described in Fig 5. Data are representative of 3 independent experiments, and n=5-11 mice per group, *P<0.05, **P<0.01, ***P<0.001. (Statistical significance calculated by ANOVA (d) or Student's t test, error bars s.e.m.)
Supplementary Figure 7 Model for the regulation of TCF-1 by SGK1.
In WT T cells, activation of mTORC2 downstream of TCR signaling results in activation of SGK1. In turn, SGK1 phosphorylates and inhibits GSK-3β. When GSK-3β is inactive, its target β-catenin cannot be degraded. Active β-catenin cooperates with the long isoforms of TCF-1 to promote transcription of TCF-1, which inhibits expression of IFN-γ. In T-Sgk1-/- cells, GSK-3β is active and thus targets β-catenin for destruction. Loss of β-catenin leads to decreased expression of TCF-1 long isoforms, and thus increased expression of IFN-γ.
Supplementary Figure 8 Diminished melanoma tumor burden in T-Sgk1-/- mice.
Representative images from one of 3 independent experiments showing reduced melanoma tumor burden in the lungs of T-Sgk1–/– mice as compared to WT mice.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–8 (PDF 1145 kb)
Rights and permissions
About this article
Cite this article
Heikamp, E., Patel, C., Collins, S. et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol 15, 457–464 (2014). https://doi.org/10.1038/ni.2867
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.2867
This article is cited by
-
The regulation and differentiation of regulatory T cells and their dysfunction in autoimmune diseases
Nature Reviews Immunology (2024)
-
Estrogen-sensitive activation of SGK1 induces M2 macrophages with anti-inflammatory properties and a Th2 response at the maternal–fetal interface
Reproductive Biology and Endocrinology (2023)
-
PCSK9 facilitates melanoma pathogenesis via a network regulating tumor immunity
Journal of Experimental & Clinical Cancer Research (2023)
-
Serum/glucocorticoid-inducible kinase 1 deficiency induces NLRP3 inflammasome activation and autoinflammation of macrophages in a murine endolymphatic hydrops model
Nature Communications (2023)
-
Role of Serum/Glucocorticoid-Regulated Kinase 1 (SGK1) in Immune and Inflammatory Diseases
Inflammation (2023)
