
Abstract
Most antigenic peptides presented by major histocompatibility complex (MHC) class I molecules are produced by the proteasome. Here we show that a proteasome-independent peptide derived from the human tumor protein MAGE-A3 is produced directly by insulin-degrading enzyme (IDE), a cytosolic metallopeptidase. Cytotoxic T lymphocyte recognition of tumor cells was reduced after metallopeptidase inhibition or IDE silencing. Separate inhibition of the metallopeptidase and the proteasome impaired degradation of MAGE-A3 proteins, and simultaneous inhibition of both further stabilized MAGE-A3 proteins. These results suggest that MAGE-A3 proteins are degraded along two parallel pathways that involve either the proteasome or IDE and produce different sets of antigenic peptides presented by MHC class I molecules.
Similar content being viewed by others


Main
Degradation of intracellular proteins is a constitutive physiological process that ensures maintenance of cellular integrity. This highly regulated process essentially occurs along two pathways: the ubiquitin-proteasome pathway and the autophagy pathway1. The proteasome is a self-compartmentalizing protease that degrades proteins tagged for degradation by covalent binding of ubiquitin. Autophagy is the degradation of cellular components in the lysosomal compartment. Peptides derived from proteins degraded by autophagy can be presented to CD4+ T lymphocytes by MHC class II molecules2, whereas peptides presented to CD8+ T lymphocytes by MHC class I molecules mainly result from cytosolic degradation of intracellular proteins by the proteasome3,4. The latter is considered as the main enzyme responsible for producing the C terminus of antigenic peptides presented by MHC class I molecules5. However, such peptides may be produced as N-terminally extended precursors that may be further cleaved by various aminopeptidases both in the cytosol and in the endoplasmic reticulum6. By studying the processing of a peptide presented by human leukocyte antigen (HLA) class I molecules, we show here that a cytosolic metallopeptidase—IDE, or insulysin—has a critical role in the production of this peptide. IDE can produce both the C terminus and the N terminus of the peptide and appears to be involved in the degradation of the parental protein.
Results
Proteasome-independent processing of a MAGE-A3 peptide
The peptide we studied is derived from MAGE-A3, a cytosolic protein expressed in human tumor cells7,8. It is presented by HLA-A1 and recognized by antitumor CD8+ cytotoxic T lymphocytes (CTLs), such as CTL clone A10, which can be isolated from individuals with cancer8,9. This peptide, which corresponds to positions 168–176 (EVDPIGHLY) of the MAGE-A3 protein, has been widely used to immunize people with melanoma in clinical trials of cancer immunotherapy10. To investigate the processing of this antigenic peptide, we examined whether recognition of tumor cells was affected by inhibition of the proteasome, which produces the majority of antigenic peptides presented to CTLs by MHC class I molecules4. We observed that proteasome-inhibited melanoma cells (CP50-MEL) were still recognized by CTL clone A10, whereas they were no longer recognized by CTL clone 246/15, which is directed against a proteasome-dependent melanoma antigen derived from Melan-A (ref. 11). This was observed using three different proteasome inhibitors: epoxomicin (Table 1), lactacystin and Ada(Ahx)3(Leu)3-vinylsulfone (data not shown).
Antigenic peptides are usually produced in the cytosol and transported into the endoplasmic reticulum by a dedicated transporter named TAP (transporter associated with antigen processing), where they associate with newly synthesized MHC class I molecules. Previous examples of proteasome-independent antigenic peptides correspond to peptides derived from the signal sequence of secretory proteins. These peptides are produced directly in the endoplasmic reticulum and therefore require neither TAP nor the proteasome for their processing12. We observed that the inhibition of TAP prevented the presentation of peptide MAGE-A3168–176 to CTL A10 (Supplementary Fig. 1), indicating that the production of this peptide occurred in the cytosol. This is in line with the lack of a signal sequence and the cytosolic location of the MAGE-A3 protein13, and it suggests that a cytosolic peptidase different from the proteasome was responsible for the processing of the MAGE-A3168–176 peptide.
A cytosolic metalloendopeptidase produces MAGE-A3168–176
Several other peptidases were found to have a role in the processing of antigenic peptides presented by class I molecules, but this role is usually limited to the trimming of the N terminus of peptide precursors produced by the proteasome14,15. The proteasome is the only cytosolic enzyme identified so far that is able to produce the C terminus of antigenic peptides5. The only exception is the tripeptidyl peptidase II (TPPII), a cytosolic aminopeptidase with some endoproteolytic activity that was found to produce two proteasome-independent viral peptides16,17. However, the role of this enzyme in antigen processing was recently questioned, and its function may be limited to N-terminal trimming or even destruction of peptides produced by the proteasome18,19. We excluded a role of TPPII in the processing of MAGE-A3168–176 by showing that CP50-MEL melanoma cells were still recognized by CTL A10 after treatment with AAF-CMK or butabindide, two inhibitors of this peptidase (Table 1). As expected, inhibitors of autophagy also failed to prevent recognition of CP50-MEL melanoma cells by CTL A10, which is in line with the fact that peptides produced in the lysosomal compartment are presented by MHC class II molecules and not by MHC class I (Table 1).
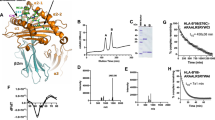
To characterize the cytosolic peptidase responsible for the production of the MAGE-A3168–176 peptide, we set up an in vitro assay using a 20-amino-acid precursor peptide containing the 9-amino-acid antigenic peptide (FGIELMEVDPIGHLYIFATS). We incubated the precursor peptide with various subcellular fractions from Epstein-Barr virus (EBV)-transformed B lymphocytes (EB81-EBV) and measured the production of the antigenic peptide by loading the digests onto target cells and evaluating their ability to activate the anti-MAGE-A3168–176 CTL clone. The antigenic peptide was detected after digestion of the precursor peptide with the cytosolic fraction but not with the nuclear fraction and not with the fraction containing the other subcellular organelles (Fig. 1a). The amount of antigenic peptide produced with the cytosolic fraction was higher than with the unfractionated postnuclear extract, which is consistent with an enrichment of the activity in the cytosolic fraction. Presence of the 9-amino-acid antigenic peptide in the digest was confirmed by high-performance liquid chromatography (HPLC) coupled to tandem mass spectrometry (MS/MS) (data not shown). To check against lysosomal enzyme contamination, we confirmed the absence of cathepsin B and cathepsin H in the cytosolic fraction (Supplementary Fig. 2). To exclude the involvement of the proteasome in this experimental setting, we either treated the cytosolic fraction with a proteasome inhibitor, depleted the fraction of proteasomes by immunoaffinity, or both, before testing its ability to produce the antigenic peptide. The production of the antigenic peptide was not affected by these treatments (Fig. 1a). The result obtained with the proteasome-depleted fraction was particularly important to definitely exclude the involvement of the proteasome because proteasome inhibitors do not block all proteasome activities and may therefore lead to erroneous interpretations20. These results confirm that a cytosolic protease, distinct from the proteasome, produces the MAGE-A3168–176 peptide.

(a) Activation of MAGE-A3–specific CTLs by digestion products resulting from incubation of precursor peptide 162-FGIELMEVDPIGHLYIFATS-181 (5 μg) with detergent-treated unfractionated postnuclear extract or subcellular fractions of EB81-EBV cells (5 μg of proteins each). The digests obtained at the indicated time points were loaded onto EB81-EBV target cells and tested for recognition by MAGE-A3–specific CTL clone 12.23. The MLP fraction contains mitochondria, lysosomes, microsomes, plasma membranes and peroxysomes. Cytosolic fractions were untreated, depleted of proteasomes by immunoaffinity, or supplemented with lactacystin (20 μM, 30 min before the precursor peptide) as indicated. (b–d) Activation of CTL clone A10 by CP50-EBV target cells loaded with digestion products obtained by incubating the precursor peptide with the cytosolic fraction of CP50-EBV cells as in a. (b) Inhibitors of cysteine peptidases (E-64, 100 μM), aspartyl peptidases (pepstatin-A, 5 μM), serine peptidases (AEBSF, 200 μM) and metallopeptidases (ortho-phenanthroline, 1 mM) were included in the digestion mixture as indicated. Inset: antigenic peptide EVDPIGHLY was incubated with the inhibitors, loaded onto target cells and tested in the same conditions as the digests, to exclude an effect of the inhibitors on CTL activity. (c) The cytosolic fraction from CP50-EBV cells was size-separated by gel filtration, and the fractions were incubated 180 min with the precursor peptide. Bottom: immunoblots of fractions using IDE- and TOP-specific antibodies. (d) The cytosolic fraction was depleted of IDE by immunoaffinity before incubation with the precursor peptide. Bottom: IDE immunoblot of the indicated fractions. Data are representative of three (a,c) or two (b,d) experiments (mean ± s.e.m. of duplicates).
We repeated the in vitro digestions of the precursor peptide, as described above, adding inhibitors of known catalytic types of peptidases, such as cysteine peptidases, aspartyl peptidases, serine peptidases and metallopeptidases. The production of the antigenic peptide was completely abolished after inhibition of metallopeptidases with ortho-phenanthroline (Fig. 1b). To confirm the involvement of a metallopeptidase in the production of the antigenic peptide in vivo, we treated CP50-MEL melanoma cells with ortho-phenanthroline and evaluated their recognition by CTLs. A clear reduction in recognition was observed with MAGE-A3–specific CTL A10, whereas recognition of the same melanoma cells by CTL clone 246/15 directed against the proteasome-dependent Melan-A antigen was not affected (Table 1).
Presentation of an antigenic peptide can also be achieved by introducing a precursor peptide into target cells by electroporation21 (Supplementary Fig. 3). In this setting also, production of the MAGE-A3168–176 peptide was blocked by treatment of target cells with ortho-phenanthroline before electroporation (Supplementary Fig. 3). The metallopeptidase family contains endopeptidases and aminopeptidases. Treatment with the aminopeptidase inhibitors bestatin and leucinethiol did not affect the production of the antigenic peptide (Supplementary Table 1 and Supplementary Fig. 3). In addition, a precursor peptide containing an amide at the C terminus and an acetyl group at the N terminus to prevent the action of exopeptidases was processed into the antigenic peptide as efficiently as the nonmodified precursor following electroporation. The processing was abolished by treatment of cells with ortho-phenanthroline (Supplementary Fig. 3). These results confirm that the enzyme producing antigenic peptide MAGE-A3168–176 is an endopeptidase of the metallopeptidase family.
Production of the MAGE-A3168–176 peptide by IDE
A limited number of cytosolic metalloendopeptidases have been described. They include thimet oligopeptidase (TOP), IDE, nardilysin and eupitrilysin22,23,24,25. Nardilysin is known to cleave at the N terminus of arginine residues in dibasic pairs26—a motif absent in the MAGE-A3 protein sequence. In addition, nardilysin is inhibited by bestatin, which did not prevent production of the MAGE-A3168–176 peptide (Supplementary Table 1)24, indicating that nardilysin is unlikely to be involved in MAGE-A3 peptide processing. Using size-exclusion chromatography, we separated the cytosolic proteins and tested the fractions for their ability to produce the antigenic peptide after incubation with the 20-mer precursor peptide. A peak of activity was detected in fractions containing proteins of about 230 kDa (Fig. 1c). As assayed by immunoblotting, these fractions contained IDE but not TOP, which was found in fractions of lower molecular weight, as expected (Fig. 1c). The involvement of TOP was also excluded by the lack of inhibitory effect of phosphoramidon or Cpp-AAF-pAB on presentation of the antigen by melanoma cells or precursor peptide–electroporated cells (Supplementary Table 1 and Supplementary Fig. 3), and because treatment of the cytosolic fraction with Cpp-AAF-pAB failed to prevent the production of the antigenic peptide (Supplementary Table 2)22,27. Although the size of IDE observed by western blot is around 115 kDa, we detected IDE, as well as the activity producing the MAGE-A3 peptide, in fractions containing proteins of 100 to 400 kDa, indicating that native IDE exists as a mixture of oligomers, as already reported28,29. These results suggested that IDE was responsible for the production of the antigenic peptide by the cytosolic fraction. This was confirmed by the observation that immunodepletion of IDE from the cytosolic fraction abolished the capacity to produce the antigenic peptide (Fig. 1d).
IDE is a ubiquitous zinc metalloendopeptidase that is also called insulysin, insulinase and insuline protease. It belongs to the inverzincin family, which is also called the M16 family in the MEROPS database23,30. IDE was initially described for its ability to degrade insulin; however, its ubiquitous expression, its predominant cytosolic location and its high degree of conservation through evolution suggest that its main function might be different from insulin degradation.
To directly assess the ability of IDE to produce the antigenic peptide, we incubated the precursor peptide with recombinant human IDE (rhIDE, more than 95% purity) and measured the production of the antigenic peptide using CTL A10. The digests were strongly recognized by CTL A10, indicating that rhIDE was able to produce the antigenic peptide in vitro (Fig. 2a). rhIDE digestions of the precursor peptide in the presence of different inhibitors resulted in an inhibition profile identical to that obtained with the cytosolic fraction—that is, the production of the antigenic peptide was sensitive to ortho-phenanthroline and N-ethylmaleimide and resistant to phosphoramidon and Cpp-AAF-pAB (Supplementary Table 2). We also analyzed the digests by HPLC-MS and identified the fragments produced (Fig. 2b). The stronger cleavage occurred at the C terminus of the antigenic peptide, as indicated by the abundance of fragment FGIELMEVDPIGHLY. The final antigenic peptide EVDPIGHLY was clearly detected in the digest (Fig. 2c), indicating that rhIDE was able to produce not only the C terminus but also the N terminus of the antigenic peptide. These results suggest that IDE might also produce the final antigenic peptide in cells—a notion in line with the failure of leucinethiol, an inhibitor of trimming aminopeptidases including ERAP, to block presentation of the antigen by tumor cells (Supplementary Table 1).

(a) Activation of CTL clone A10 by digestion products resulting from incubation of precursor peptide FGIELMEVDPIGHLYIFATS with recombinant human IDE. The digests obtained at the indicated time points were loaded onto CP50-EBV target cells and tested for recognition by CTL clone A10. (b) Peptide fragments detected by HPLC-MS analysis of a digest obtained as in a after 60 min of incubation (85% of the precursor peptide degraded). Top: horizontal lines indicate peptide fragments; thickness indicates the abundance of the fragment. Numbers above the lines indicate MS estimates of the amount of each fragment in arbitrary units. Arrows indicate cleavage sites. The sequence of the antigenic peptide is written in bold. A similar pattern was observed at various digestion times. Bottom: reconstructed ion current (RIC) chromatograms for the HPLC-MS analysis of the same digest. The digest was separated by HPLC and analyzed on-line by MS. The amount of peptide fragments was estimated by integrating the area of the relevant peaks on chromatograms plotting the current measured for ions with relevant m/z. Fragments with a methionine were estimated by summing the signals of ions with an oxidized methionine (m) and those of ions with a non-oxidized methionine (M). The ordinate is normalized to the most abundant peptide fragment (FGIELMEVDPIGHLY). (c) Detection of the antigenic peptide by HPLC-MS/MS in the same digest as in b. The fragments detected by MS/MS are indicated above the peptide sequence for N-terminal b-H2O ions and below the sequence for C-terminal y ions. Top: RIC chromatogram for the HPLC-MS/MS analysis of the digest. The chromatogram was obtained by plotting the current measured for ions with m/z 536 and 489 obtained after MS/MS analysis of ion with m/z 521.8, which corresponds to the doubly charged ion of EVDPIGHLY. These two ions are the most abundant ions obtained after fragmentation of the synthetic antigenic peptide, as shown in the bottom panel. AU, arbitrary units. Middle: fragmentation spectrum of the doubly charged ion with m/z 521.82+ detected in the digest between retention times 8.5 and 9.1 min. Bottom: fragmentation spectrum of the synthetic antigenic peptide EVDPIGHLY. Ion with m/z 5132+ is the dehydrated derivative of precursor ion with m/z 521.82+. Data are representative of three experiments (mean ± s.e.m. of duplicates).
To confirm the involvement of IDE in the production of peptide MAGE-A3 by tumor cells, we used RNA interference to reduce expression of IDE in tumor cells. We electroporated IDE-specific RNAi into HLA-A1+ tumor cells that do not express MAGE-A3. Three days later, at the time of lowest IDE expression, we infected the electroporated cells with a recombinant vaccinia virus encoding MAGE-A3, and we tested the cells for recognition by CTL A10 in a lysis assay. We observed a clear reduction in CTL recognition of IDE RNAi–treated cells compared to control RNAi–treated cells (Fig. 3a). A parallel reduction of IDE protein expression was observed by immunoblot in IDE RNAi–treated cells (Fig. 3b). The remaining CTL recognition of IDE RNAi–transfected cells resulted presumably from residual IDE expression, as it was further reduced by ortho-phenanthroline treatment but not by epoxomicin. These results confirm the role of IDE in the production of MAGE-A3168–176 in tumor cells.

(a) Lysis by CTL clone A10 of HT-29 colon carcinoma cells (HLA-A1+) transfected with IDE-specific or control (ctrl) siRNA and infected 72 h later with a recombinant vaccinia virus encoding MAGE-A3. Epoxomicin (1 μM) and ortho-phenanthroline (1.5 mM) were added as indicated. Treated cells pulsed with peptide EVDPIGHLY (5 μg ml−1) were also tested to check viability. Chromium release was measured after 4 h. (b) Immunoblot detection of IDE and MAGE-A3 in an aliquot of cells from a. Loading control: α-tubulin. Data are representative of nine experiments, two of which included epoxomicin and ortho-phenanthroline (mean ± s.e.m. of duplicates).
IDE is involved in MAGE-A3 protein turnover
The MAGE-A3 protein comprises other antigenic peptides, and the processing of at least one of them, MAGE-A3114–122 presented by HLA-B40, depends on the proteasome31. Metallopeptidase inhibition did not prevent presentation of this peptide by CP50-MEL melanoma cells, as opposed to proteasome inhibition, which indicates that IDE is not involved in the processing of MAGE-A3114–122 (Supplementary Fig. 4). The involvement of two distinct proteases in the production of two antigenic peptides from the MAGE-A3 protein suggests that both proteases have a role in the breakdown of this protein. Their roles could be sequential (one protease acting on the products of the other) or parallel (each protease being involved in the degradation of a fraction of the MAGE-A3 proteins). To distinguish between these possibilities, we studied the degradation of the MAGE-A3 protein in CP50-MEL melanoma cells treated with epoxomicin (to block the proteasome) or with ortho-phenanthroline (to block IDE) (Fig. 4). MAGE-A3 protein degradation was already clear 8 h after blocking protein synthesis with cycloheximide and was complete after 16 h. It was partly prevented by the addition of epoxomicin but also by the addition of ortho-phenanthroline. In contrast, the degradation of melanocytic protein Melan-A, and that of p21, a known proteasome substrate, were prevented by epoxomicin but not by ortho-phenanthroline. These results confirmed the role of a metallopeptidase, most likely IDE, in MAGE-A3 protein degradation. Moreover, an additive effect was observed when epoxomicin and ortho-phenanthroline were added together, resulting in complete protection of MAGE-A3 from degradation. Identical results were obtained using another melanoma cell line: CP64-MEL (Supplementary Fig. 5).

Immunoblot analysis of MAGE-A3 expression in lysates of CP50-MEL melanoma cells treated for 1 h with cycloheximide (100 μg ml−1), epoxomicin (1.5 μM) and ortho-phenanthroline (1 mM) as indicated, washed and incubated for the indicated time. MAGE-A3 signals were corrected based on the loading controls (α-tubulin, β-actin and IDE) and expressed in % relative to the untreated condition. Data are representative of seven experiments, four of which included Melan-A and p21 (controls).
Because ortho-phenanthroline blocks not only IDE but also other metallopeptidases, we evaluated MAGE-A3 protein turnover in IDE RNAi–silenced human embryonic kidney (HEK) 293 cells. Although IDE silencing was not optimal in these cells, the MAGE-A3 protein was partly protected from degradation, which is in line with the notion that the effect of ortho-phenanthroline in the above experiments was due to IDE inhibition (Fig. 5). Problems with cell viability prevented us from testing proteasome inhibitors in combination with IDE RNAi. However, these results indicate that IDE and the proteasome can act in two parallel pathways for degradation of the MAGE-A3 protein.

Immunoblot analysis of MAGE-A3 and IDE expression in HEK 293 cells stably expressing MAGE-A3 and transfected with IDE-specific or control siRNA. Protein expression was analyzed 16 h after cycloheximide treatment (100 μg ml−1) for 1 h to inhibit protein synthesis. Loading control: α-tubulin. Data are representative of two experiments.
Discussion
Although IDE was initially described for its ability to degrade insulin, its actual involvement in insulin turnover is debated, mostly because of its predominant cytosolic location32. It was suggested that IDE might be part of a second cytosolic protein breakdown pathway, with preference for substrates that are not efficiently targeted to or degraded by the proteasome33. Several IDE substrates were described, including glucagon, atrial natriuretic peptide, transforming growth factor-α, amyloid-β peptide and oxidized proteins23,34,35,36. The observation that IDE can degrade amyloid-β peptide stimulated numerous studies looking for a potential link between IDE activity and Alzheimer's disease. Genetic studies implicate the IDE locus in susceptibility to both Alzheimer's disease and type 2 diabetes mellitus37. Accordingly, IDE-deficient mice show accumulation of amyloid-β protein, hyperinsulinemia and glucose intolerance38.
Structural studies have indicated that the IDE catalytic site accommodates substrates of relatively small size within a pocket that shifts from an open conformation, to allow for substrate entry, to a closed conformation required for optimal catalysis39. However, large oxidized proteins are degraded by IDE, suggesting that the basal activity of the IDE catalytic pocket in the open conformation suffices to degrade such substrates35,36. Whether the full-length MAGE-A3 protein is directly taken up by IDE or whether other enzymes are involved remains to be established.
The cleavage specificity of IDE is poorly defined, and known substrates are diverse in sequence and structure. Structural studies of IDE-substrate complexes suggest that positively charged residues at the C terminus of the substrate prevent its interaction with IDE39. These studies also showed that substrates bound in the catalytic pocket of IDE locally adopt a β-sheet secondary structure39. A common feature of many IDE substrates is a tendency to form amyloid fibrils, and IDE was therefore suggested to act in the degradation of amyloidogenic proteins, possibly by recognizing secondary structures enriched in β-sheets40. A function of IDE might be to protect the cell from aggregation of such substrates in amyloid deposits. Because the structure of MAGE-A3 has not been solved, it is difficult to determine whether such features apply to MAGE-A3.
Our data implicate IDE in the production of antigenic peptides, exemplified by the MAGE-A3 peptide presented by HLA-A1. A fraction of the MAGE-A3 proteins is probably degraded by the proteasome, resulting in the production of one set of antigenic peptides, and another fraction is degraded by IDE, contributing to a second set of antigenic peptides. Inhibitors of autophagy, the other main pathway for degradation of intracellular proteins, did not block MAGE-A3 protein degradation or presentation of MAGE-A3168–176. Notably, IDE was able to produce both the C terminus and the N terminus of this antigenic peptide, which therefore does not need N-terminal trimming. Whether IDE produces many other antigenic peptides and contributes significantly to the class I peptide repertoire remains to be established.
Besides the proteasome, the only protease found able to produce the C terminus of an antigenic peptide is TPPII, which was shown to produce two viral peptides16,17. These results initiated large efforts to study the role of TPPII in antigen processing. Although no further evidence for a role of TPPII in C-terminal processing has emerged, TPPII was found to contribute to N-terminal trimming of N-extended peptide precursors18,41. Such extended precursors can be produced by the proteasome and need to be trimmed to achieve their final length. This trimming may occur in the endoplasmic reticulum through the action of dedicated aminopeptidases called ERAP1 and ERAP2 (refs. 6,14,15). Trimming in the endoplasmic reticulum is thought to allow the presentation of certain peptides, such as those with a proline in position 2, which are more efficiently transported by TAP as N-extended precursors than in their final size. In other cases, the N-extended precursor may be trimmed in the cytosol by apparently redundant aminopeptidases including TPPII, leucine aminopeptidase, bleomycin hydrolase and puromycin-sensitive aminopeptidase6. Free peptides are potentially harmful within the cytosol; they have a very short half-life, most of them being rapidly cleaved by a number of cytosolic peptidases, including to a certain extent some of those mentioned above6. A major peptidase involved in peptide destruction is TOP—one of the few other cytosolic metallopeptidases. Reducing TOP expression with siRNA was found to enhance presentation of antigenic peptides42. Cytosolic destruction of antigenic peptides therefore competes with their production and contributes to the generally low efficiency of class I antigen processing.
In conclusion, this report describes a new role for IDE in the production of antigenic peptides presented by MHC class I molecules. Our data also suggest a new substrate for IDE and point to a role for IDE in degrading some intracellular proteins, indicating the presence, besides the proteasome, of a second nonlysosomal pathway for degradation of intracellular proteins, with IDE as the responsible protease. This pathway may be particularly suited to degradation of proteins that are poor proteasome substrates and/or tend to form aggregates or amyloid deposits. Degradation of intracellular proteins by IDE would occur along a pathway parallel to that of the proteasome. Because of their different specificity, IDE and the proteasome would produce a different set of peptides, which together contribute to the large diversity of peptides displayed at the cell surface by MHC class I molecules for scrutiny by CD8 T lymphocytes.
Methods
Cellular reagents and cell culture.
Melanoma cell line CP50-MEL and EBV-transformed B cell line CP50-EBV were obtained from HLA-A1 melanoma patient CP50. EBV-transformed B cell line EB81-EBV was obtained from HLA-A1 melanoma patient EB81. All human samples were obtained from patients who provided informed consent, and the experiments were approved by the Commission d'Ethique biomédicale hospitalo-facultaire of the Université catholique de Louvain. These cell lines were grown in Iscove's medium (Invitrogen) supplemented with 10% (vol/vol) FCS (Hyclone), amino acids and antibiotics. Colon carcinoma cell line HT-29 was obtained from ATCC and was grown in DMEM (Invitrogen) supplemented as above. MAGE-A3168–176–specific CTL clone LB1965-659/12.23, which was used in Figure 1a, was isolated as described10. In all the other experiments, we used the MAGE-A3168–176–specific CTL clone LAU147-810/A10 (called A10 throughout), which had been isolated from an HLA-A1 melanoma patient vaccinated with a recombinant canarypox virus encoding the MAGE-A3168–176 peptide. Melan-A–specific CTL clone 246/15 and tyrosinase-specific CTL clones IVSB and 210/9 were described11,12. CTL clones were stimulated every 3 to 4 weeks with 3 × 105 irradiated (100 Gy) CP50-EBV cells pulsed with 5 μg ml−1 of antigenic peptides for 30 min, 106 irradiated LG2-EBV cells as feeder cells, human recombinant IL-2 (50 U ml−1), and the indoleamine 2,3-dioxygenase inhibitor 1-methyl-L-tryptophan (200 μM, Sigma) in Iscove's medium with 10% (vol/vol) human serum43. The recombinant MAGE-A3 vaccinia virus was provided by V. Cerundolo (University of Oxford).
Reagents.
All inhibitors were from Sigma, except butabindide (Tocris), Cpp-AAF-pAB (Bachem) and lactacystin (Biomol Int.). Recombinant interferon-γ was from Preprotech. All antibodies used were mouse monoclonal antibodies: IDE (clone 9B12.221, Covance, further purified on HiTrap protein G), TOP (4D6, Upstate), p21 (2946-1, Cell Signaling), Melan-A/MART-1 (A103, Dako), β-actin (AC-15, Sigma), α-tubulin (58666, Santa Cruz). The monoclonal antibody against MAGE-A3 (57B) was provided by G. Spagnoli (University of Basel)44. Horseradish peroxidase (HRP)-linked goat anti-mouse IgG antibodies (2302) were from Santa Cruz. Peptides were synthesized on solid phase using Fmoc chemistry and purified by reversed phase HPLC, lyophilized and conserved in DMSO at 10–20 mg ml−1. Recombinant human IDE, which was produced in insect cell line Sf21, was more than 95% pure (R&D Systems).
TAP inhibition.
ICP-47 cloned in PBJ1-Neo was a gift from H.-G. Rammensee (University of Tübingen). 293-EBNA cells (Invitrogen) were transiently transfected with MAGE-A3 (cloned in pEF-BOS-puro), tyrosinase (pCDNA3), HLA-A1 (pCDNA1), HLA-A2.1 (pCDNA3) and ICP47 using lipofectamin (Invitrogen). We resuspended cells 24 h later in complete medium with β2-microglobulin (2.5 μg ml−1) and used them for the CTL assay.
Protease inhibition in CP50-MEL melanoma cells.
For proteasome inhibition, cells were incubated 1 h with 0.06 to 1 μM epoxomicin, then overnight with 0.012 μM epoxomicin. We loaded half of them with the antigenic peptide (2 μg ml−1) in the presence of β2-microglobulin (2 μg ml−1) for 30 min before the CTL assay. For metallopeptidase inhibition, we treated cells overnight with ortho-phenanthroline (25 to 200 μM). Half of them were loaded with the antigenic peptide (5 μg ml−1, 1 h) before the CTL assay. For treatments with AAF-CMK, butabindide, bestatin, phosphoramidon, Cpp-AAF-pAB, leupeptin, E64d, PMSF and pepstatin-A, we first treated cells at pH 3 for 3 min at 25 °C with a glycine buffer to elute MHC-bound peptides, and then we incubated them overnight with the lowest concentration of inhibitor indicated in the table, in Iscove's medium without FCS. Three hours before the CTL assay, we added the actual concentration of inhibitor as indicated in the table. For treatment with leucinethiol, cells were acid-eluted as above and treated for 5 h with the indicated concentration of leucinethiol (stock solution containing 1 mM Tris (2-carboxyethyl)phosphine). For treatment with 3-methyladenine, bafilomycine-A1 and chloroquine, cells were acid-eluted as above, then treated 1 h with the dose indicated in Table 1 and further incubated overnight with the lowest dose indicated in Table 1. Half of the cells were loaded with the antigenic peptide (2 μg ml−1) 30 min before the CTL assay. For the CTL assay, we incubated cells (20–30,000 per well) with the relevant CTL clone (10,000 cells per well) in the presence of 25 U ml−1 IL-2, and we measured the production of IFN-γ by ELISA (Biosource-Invitrogen) after an overnight incubation.
Subcellular fractionation.
We homogenized 150–200 × 106 cells from line CP50-EBV or EB81-EBV (MAGE-A3 negative) with a Dounce in sucrose 250 mM and imidazole 3 mM pH 6.9, as described45, except that we omitted EDTA to prevent inhibition of metallopeptidases. The postnuclear extract was separated from nuclei and cell debris (nuclear fraction) by four cycles of homogenization and centrifugation. The postnuclear extract was then centrifuged (112,000g, 1 h, 4 °C) to separate the cytosolic fraction (nonprecipitable supernatant) from the MLP pellet. In the initial screening, we tested the fractions in the presence of 0.5% (vol/vol) Triton X-100 to liberate the content of the organelles.
Assay for the peptidase activity producing peptide MAGE-A3168–176.
The 20-mer precursor peptide (FGIELMEVDPIGHLYIFATS, positions 162–181, where cysteine 181 is replaced by serine to prevent dimerization) (0.25 μg μl−1) was incubated at 37 °C with the cytosolic fraction (0.25 μg μl−1 proteins) in 140–200 μl of Tris 50 mM, NaCl 100 mM, pH 7.5. We collected aliquots (20 μl) at different time points and stopped the reaction with 5% (wt/vol) trichloroacetic acid (final concentration). Proteins were precipitated by centrifugation (20,000g, 20 min, 4 °C). Supernatants were lyophilized and resuspended in 55 μl serum-free medium (X-vivo 10, Invitrogen). We loaded 25 μl in duplicates on CP50-EBV cells (40,000 per microwell) during 30 min. We added CTL A10 (10,000 cells per well in 25 U ml−1 IL-2) and measured IFN-γ production after an overnight incubation. The presence of antigenic peptide EVDPIGHLY was confirmed by mass spectrometry.
Depletions.
For proteasome depletion, we incubated the cytosolic fraction (1 ml, 4 h, 4 °C) with CNBr-activated sepharose beads (100 μl) saturated with monoclonal antibody MCP-21 directed against the α2 subunit of the proteasome. The efficiency of depletion was estimated to be between 75% and 87% using a quantitative ELISA as described31. For IDE depletion, the cytosolic fraction (180 μl) was incubated (2 h, 4 °C) with protein G–sepharose beads (Amersham) saturated with purified anti-IDE monoclonal antibody (80 μl). The supernatant was recovered after filtration on a Nylon membrane (10 μm). We analyzed the efficiency of IDE depletion by western blot (Nu-PAGE 15% Bis-Tris; nitrocellulose Hybond-C extra; semi-dry transfer). The membrane was saturated (1 h, phosphate-buffered saline, powdered skim milk (5% (wt/vol)), Tween 20 (0.1% (vol/vol))), incubated with anti-IDE purified monoclonal antibody (1/1000) and revealed with HRP-linked goat anti-mouse IgG antibodies (1/1000). We digested the 20-amino-acid precursor peptide (5 μg per time point) with 156 μl of depleted fractions, and we took aliquots after 0, 30, 60 and 180 min.
Gel filtration.
A cytosolic fraction of CP50-EBV (25 mg proteins) was precipitated with 40% (vol/vol) of a saturated (4 °C) ammonium sulfate solution, and the supernatant was concentrated with a second precipitation at 80% (vol/vol) of the same ammonium sulfate solution. We resuspended the pellet with the equilibrating buffer of the gel filtration (Tris 50 mM, NaCl 100 mM, pH 7.4). The separation was done on a Superdex 200 HR 10/30 (Amersham) with a flow rate of 0.5 ml min−1, and fractions (0.5 or 1 ml) were collected between 3 ml and 21 ml of retention volume. We performed a digestion of the 20-mer precursor peptide (0.3 μg μl−1) with each fraction (100 μl) at 37 °C during 3 h. The production of the MAGE-A3168–176 antigenic peptide was evaluated in a CTL assay. We analyzed 20 μl of the fractions between 6 and 17 ml of retention volume by immunoblot with IDE monoclonal antibody (1/1000), and we simultaneously added TOP monoclonal antibody (1 μg ml−1).
Degradation of the precursor peptide by recombinant IDE.
We digested the 20-mer precursor peptide (0.25 μg μl−1) with rhIDE (2.5 ng μl−1) in 140–200 μl of Tris 50 mM, NaCl 100 mM pH 7.5. Aliquots of 20 μl were collected and stopped on dry ice. We added 35 μl of X-vivo medium, and we performed the CTL assay as above. For the mass spectrometry (MS) analysis, aliquots were injected on a reverse-phase chromatography PepMap C18 0.3/15 (LC Packings) and eluted with a 35 min linear gradient of acetonitrile in water (5–50% (vol/vol)) containing 0.05% (vol/vol) trifluoroacetic acid with a flow rate of 4 μl min−1. The MS analysis was performed on-line with an LCQ Deca XP ion-trap spectrometer equipped with an electrospray ionization source (ThermoFinnigan).
Peptide electroporation.
We treated (or did not treat) CP50-EBV cells (1.2 × 107) for 2 h before the electroporation with o-phenanthroline (1 mM), bestatin (750 μM), Cpp-AAF-pAB (250 μM) or a mix of lactacystin and butabindide (50 μM each) in medium X-vivo 10. Cells were washed with phosphate-buffered saline, resuspended in 2 × 400 μl of cytoporation buffer (Cyto Pulse Sciences Inc.) and transferred into 0.4 cm Genepulser cuvettes (Biorad), and peptides (100 μM) were electroporated (10 pulses, 5 ms, 480 V, 1 Hz). Cells were diluted in 6 ml of X-vivo 10 containing (or not) ortho-phenanthroline (25 μM), bestatin (10 μM), Cpp-AAF-pAB (10 μM) or a mix of lactacystin and butabindide (1 μM each). After 4 h, cells were washed (X-vivo 10) and used for the CTL assay.
Silencing of IDE by siRNA.
We electroporated HT-29 cells (1.6 × 106, 300 V, 150 μF, 90 ohms) with a pool of IDE-specific siRNA duplexes (50 nM each, IDE-HSS105-178, IDE-HSS105-179, IDE-HSS105-180, Invitrogen) or with control siRNA (150 nM, nontargeting control D-001210-01-20, Dharmacon) using gene pulser Xcell (Biorad). Cells were incubated 72 h in complete medium, labeled with 51Cr for 1 h and infected with a recombinant vaccinia virus encoding MAGE-A3 (MOI: 25) for 2 h. Where indicated, ortho-phenanthroline (1.5 mM) or epoxomicin (1 μM) was added 30 min before the end of infection. Cells were washed and plated for a lysis assay using CTL clone A10. In parallel, we loaded cells with the antigenic peptide (5 μg ml−1) and tested them similarly. An aliquot of target cells was collected before the CTL assay and tested by immunoblot with IDE monoclonal antibody (1/500) and MAGE-A3 monoclonal antibody (1/300) added simultaneously. Membranes were then acid-stripped (Restore buffer, Thermo) and probed with anti-α-tubulin monoclonal antibody (0.1 μg ml−1). Analysis of MAGE-A3 protein degradation in IDE-silenced cells was performed in HEK 293 stable transfectants expressing MAGE-A3, which were treated with siRNA as above, incubated 65 h, then treated 1 h with cycloheximide (100 μg ml−1), washed and analyzed by immunoblot 16 h later.
Study of MAGE-A3 degradation in melanoma cells.
We treated CP50-MEL or CP64-MEL cells for 1 h with cycloheximide (100 μg ml−1) in X-vivo 10 with or without epoxomicin (1.5 μM) and ortho-phenanthroline (1 mM). After washing, cells were incubated 4, 8 or 16 h in complete culture medium, then analyzed by immunoblot (Nu-PAGE 4–12% Bis-Tris) sequentially with monoclonal antibodies as follows: MAGE-A3 (1/300), p21 (1/800), Melan-A (25 μg ml−1), then a mix of IDE, α-tubulin and β-actin antibodies (1/800, 0.1 μg ml−1, 0.2 μg ml−1), all revealed with anti-mouse IgG-HRP antibodies (1/2000). Note that the anti-MAGE-A3 antibody used also recognizes other members of the MAGE-A family, some of which may contribute to the signal measured46. Chemiluminesence was detected using G:BOX BioImaging System. Signals were quantified using Syngene Gene Tools software and analyzed as follows. In each blot, different volumes of the MAGE-A3–positive sample from cells treated with epoxomicin and ortho-phenanthroline were loaded. Signals of each sample were reported on this curve using the corresponding equation, so as to determine a fold increase or decrease compared to the MAGE-A3–positive sample. α-tubulin, β-actin and IDE were used as loading controls because their half-lives largely exceeded the duration of the experiment. The signals obtained with each loading control were quantified and corrected similarly by reference to the dilution curve of the same sample as above. Each calculated value for MAGE-A3 was then converted to a ratio relative to the level of corrected loading control α-tubulin, β-actin or IDE in the same sample. The level of MAGE-A3 in each condition was finally expressed as a percentage relative to the MAGE-A3 level in the untreated sample.
References
Knecht, E. et al. Intracellular protein degradation in mammalian cells: recent developments. Cell. Mol. Life Sci. 66, 2427–2443 (2009).
Vyas, J.M., Van der Veen, A.G. & Ploegh, H.L. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 8, 607–618 (2008).
Rock, K.L. & Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 17, 739–779 (1999).
Shastri, N., Schwab, S. & Serwold, T. Producing nature's gene-chips: the generation of peptides for display by MHC class I molecules. Annu. Rev. Immunol. 20, 463–493 (2002).
Craiu, A., Akopian, T., Goldberg, A. & Rock, K.L. Two distinct proteolytic processes in the generation of a major histocompatibility complex class I-presented peptide. Proc. Natl. Acad. Sci. USA 94, 10850–10855 (1997).
Rock, K.L., Farfan-Arribas, D.J. & Shen, L. Proteases in MHC class I presentation and cross-presentation. J. Immunol. 184, 9–15 (2010).
van der Bruggen, P. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 (1991).
Gaugler, B. et al. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J. Exp. Med. 179, 921–930 (1994).
Boon, T., Coulie, P.G., Van den Eynde, B. & van der Bruggen, P. Human T cell responses against melanoma. Annu. Rev. Immunol. 24, 175–208 (2006).
Lonchay, C. et al. Correlation between tumor regression and T cell responses in melanoma patients vaccinated with a MAGE antigen. Proc. Natl. Acad. Sci. USA 101, 14631–14638 (2004).
Morel, S. et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity 12, 107–117 (2000).
Wölfel, C. et al. Transporter (TAP)- and proteasome-independent presentation of a melanoma-associated tyrosinase epitope. Int. J. Cancer 88, 432–438 (2000).
Kocher, T. et al. Identification and intracellular location of MAGE-3 gene product. Cancer Res. 55, 2236–2239 (1995).
Hammer, G.E., Kanaseki, T. & Shastri, N. The final touches make perfect the peptide-MHC class I repertoire. Immunity 26, 397–406 (2007).
Saveanu, L., Carroll, O., Hassainya, Y. & van Endert, P. Complexity, contradictions, and conundrums: studying post-proteasomal proteolysis in HLA class I antigen presentation. Immunol. Rev. 207, 42–59 (2005).
Seifert, U. et al. An essential role for tripeptidyl peptidase in the generation of an MHC class I epitope. Nat. Immunol. 4, 375–379 (2003).
Guil, S. et al. Need for tripeptidyl-peptidase II in major histocompatibility complex class I viral antigen processing when proteasomes are detrimental. J. Biol. Chem. 281, 39925–39934 (2006).
van Endert, P. Role of tripeptidyl peptidase II in MHC class I antigen processing - the end of controversies? Eur. J. Immunol. 38, 609–613 (2008).
Kawahara, M. et al. Analysis of the role of tripeptidyl peptidase II in MHC class I antigen presentation in vivo. J. Immunol. 183, 6069–6077 (2009).
Princiotta, M.F. et al. Cells adapted to the proteasome inhibitor 4-hydroxy- 5-iodo-3-nitrophenylacetyl-Leu-Leu-leucinal-vinyl sulfone require enzymatically active proteasomes for continued survival. Proc. Natl. Acad. Sci. USA 98, 513–518 (2001).
Vigneron, N. et al. An antigenic peptide produced by peptide splicing in the proteasome. Science 304, 587–590 (2004).
Saric, T. et al. Major histocompatibility complex class I-presented antigenic peptides are degraded in cytosolic extracts primarily by thimet oligopeptidase. J. Biol. Chem. 276, 36474–36481 (2001).
Duckworth, W.C., Bennett, R.G. & Hamel, F.G. Insulin degradation: progress and potential. Endocr. Rev. 19, 608–624 (1998).
Chesneau, V. et al. Isolation and characterization of a dibasic selective metalloendopeptidase from rat testes that cleaves at the amino terminus of arginine residues. J. Biol. Chem. 269, 2056–2061 (1994).
Mzhavia, N. et al. Cloning, expression, and characterization of human metalloprotease 1: a novel member of the pitrilysin family of metalloendoproteases. DNA Cell Biol. 18, 369–380 (1999).
Chow, K.M. et al. Nardilysin cleaves peptides at monobasic sites. Biochemistry 42, 2239–2244 (2003).
Lessley, B.A. & Garner, D.L. Purification and characterization of Pz-peptidase B, a neutral metalloendopeptidase from bovine spermatozoa. Biol. Reprod. 43, 643–658 (1990).
Safavi, A., Miller, B.C., Cottam, L. & Hersh, L.B. Identification of gamma-endorphin-generating enzyme as insulin-degrading enzyme. Biochemistry 35, 14318–14325 (1996).
Song, E.S., Juliano, M.A., Juliano, L. & Hersh, L.B. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. J. Biol. Chem. 278, 49789–49794 (2003).
Rawlings, N.D., Morton, F.R. & Barrett, A.J. MEROPS: the peptidase database. Nucleic Acids Res. 34, D270–D272 (2006).
Schultz, E.S. et al. The production of a new MAGE-3 peptide presented to cytolytic T lymphocytes by HLA-B40 requires the immunoproteasome. J. Exp. Med. 195, 391–399 (2002).
Authier, F., Posner, B.I. & Bergeron, J.J. Insulin-degrading enzyme. Clin. Invest. Med. 19, 149–160 (1996).
Kirschner, R.J. & Goldberg, A.L. A high molecular weight metalloendoprotease from the cytosol of mammalian cells. J. Biol. Chem. 258, 967–976 (1983).
Kurochkin, I.V. & Goto, S. Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 345, 33–37 (1994).
Morita, M. et al. Insulin-degrading enzyme exists inside of rat liver peroxisomes and degrades oxidized proteins. Cell Struct. Funct. 25, 309–315 (2000).
Fagan, J.M. & Waxman, L. Purification of a protease in red blood cells that degrades oxidatively damaged haemoglobin. Biochem. J. 277, 779–786 (1991).
Bertram, L. et al. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science 290, 2302–2303 (2000).
Farris, W. et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 100, 4162–4167 (2003).
Shen, Y., Joachimiak, A., Rosner, M.R. & Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443, 870–874 (2006).
Kurochkin, I.V. Insulin-degrading enzyme: embarking on amyloid destruction. Trends Biochem. Sci. 26, 421–425 (2001).
Kawahara, M. et al. Analysis of the role of tripeptidyl peptidase II in MHC class I antigen presentation in vivo. J. Immunol. 183, 6069–6077 (2009).
York, I.A. et al. The cytosolic endopeptidase, thimet oligopeptidase, destroys antigenic peptides and limits the extent of MHC class I antigen presentation. Immunity 18, 429–440 (2003).
Uyttenhove, C. et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 9, 1269–1274 (2003).
Rimoldi, D. et al. Anti-MAGE-3 antibody 57B and anti-MAGE-1 antibody 6C1 can be used to study different proteins of the MAGE-A family. Int. J. Cancer 86, 749–751 (2000).
de Diesbach, P. et al. Identification, purification and partial characterisation of an oligonucleotide receptor in membranes of HepG2 cells. Nucleic Acids Res. 28, 868–874 (2000).
Landry, C. et al. Monoclonal antibody 57B stains tumor tissues that express gene MAGE-A4. Int. J. Cancer 86, 835–841 (2000).
Acknowledgements
We thank F. Piette for technical assistance, P. van der Bruggen for suggestions, T. Boon and P. Coulie for critical reading of the manuscript, V. Cerundolo (Molecular Immunology Group, University of Oxford) for providing vaccinia virus constructs and S. Depelchin and J. Klein for editorial assistance. We thank G. Spagnoli (University of Basel) for the monoclonal antibody against MAGE-A3 (57B), and we thank H.-G. Rammensee (University of Tübingen) for the ICP-47 cloned in PBJ1-Neo. Supported by grants from the European Union under the Sixth Programme (CancerImmunotherapy; LSHC-CT-2006-518234), the Belgian Programme on Interuniversity Poles of Attraction initiated by the Belgian State (Prime Minister's Office, Science Policy Programming), the Fonds National de la Recherche Scientifique (FNRS, Belgium), the Fondation contre le Cancer (nonprofit organization, Belgium), the Fonds J. Maisin (Belgium) and the Fondation Salus Sanguinis (Belgium). N.P. was supported by a fellowship from the Fonds pour la Recherche dans l'Industrie et l'Agriculture (Belgium) and by the Télévie-FNRS (Belgium). P.v.E. was supported by grant 05-BLAN-0612 from the Agence Nationale de la Recherche (France).
Author information
Authors and Affiliations
Contributions
N.P., V.S., D.C., P.d.D., S.M., J.C., P.v.E. and B.J.V.d.E. designed the experiments. N.P., V.S., D.C., P.d.D., S.M. and J.C. performed the experiments. N.P., V.S., S.M. and B.J.V.d.E. analyzed and interpreted the data. N.P., V.S. and B.J.V.d.E. wrote the manuscript and prepared the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 and Supplementary Tables 1 and 2 (PDF 1723 kb)
Rights and permissions
About this article
Cite this article
Parmentier, N., Stroobant, V., Colau, D. et al. Production of an antigenic peptide by insulin-degrading enzyme. Nat Immunol 11, 449–454 (2010). https://doi.org/10.1038/ni.1862
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.1862
This article is cited by
-
ERAP1 promotes Hedgehog-dependent tumorigenesis by controlling USP47-mediated degradation of βTrCP
Nature Communications (2019)
-
The insulin-degrading enzyme is an allosteric modulator of the 20S proteasome and a potential competitor of the 19S
Cellular and Molecular Life Sciences (2018)
-
Towards a systems understanding of MHC class I and MHC class II antigen presentation
Nature Reviews Immunology (2011)
-
Post-proteasomal and proteasome-independent generation of MHC class I ligands
Cellular and Molecular Life Sciences (2011)
-
Providing ligands for MHC class I molecules
Cellular and Molecular Life Sciences (2011)
