
Abstract
Hirschsprung disease (HSCR), the most common hereditary cause of intestinal obstruction, shows considerable variation and complex inheritance. Coding sequence mutations in RET, GDNF, EDNRB, EDN3 and SOX10 lead to long-segment (L-HSCR) and syndromic HSCR but fail to explain the transmission of the much more common short-segment form (S-HSCR). We conducted a genome scan in families with S-HSCR and identified susceptibility loci at 3p21, 10q11 and 19q12 that seem to be necessary and sufficient to explain recurrence risk and population incidence. The gene at 10q11 is probably RET, supporting its crucial role in all forms of HSCR; however, coding sequence mutations are present in only 40% of linked families, suggesting the importance of noncoding variation. Here we show oligogenic inheritance of S-HSCR, the 3p21 and 19q12 loci as RET-dependent modifiers, and a parent-of-origin effect at RET. This study demonstrates by a complete genetic dissection why the inheritance pattern of S-HSCR is nonmendelian.
Similar content being viewed by others


Main
The molecular dissection of 'model' disorders is crucial to the elucidation of complex patterns of inheritance, which have been difficult to assess by other means1,2,3. HSCR is a neurocristopathy characterized by the absence of ganglion cells in the myenteric and submucosal plexuses of the gastrointestinal tract4. The phenotype usually occurs in isolation and is classified by extent of aganglionosis into long-segment (L-HSCR, 20% of affected individuals) and short-segment (S-HSCR, 80%) forms, each with distinct genetic characteristics5. HSCR is hereditary but nonmendelian with risk varying by gender, segment length and co-occurrence of non-enteric phenotypes6. We have previously concluded that RET is the major gene involved in HSCR, for several reasons: (i) only one affected family unlinked to RET has been reported7; (ii) coding sequence mutations occur in 50% of familial and 15–35% of sporadic cases8; (iii) even when the major mutation is in EDNRB, RET variants make some contribution to susceptibility9 and (iv) homozygous Ret-null mice have full sex-independent penetrance of aganglinonosis10. Yet, RET mutations may not be sufficient to lead to aganglionosis, as the penetrance of mutant alleles is 65% in males and 45% in females8. RET is the major gene underlying HSCR primarily in families enriched for L-HSCR5. The genes critical for the complex inheritance of the much more common S-HSCR remain unknown.
We carried out a genome search of the number and locations of genes conferring susceptibility to S-HSCR in 49 affected families ascertained through a proband with S-HSCR, using 371 tandem repeat polymorphisms at a map resolution of approximately 10 cM. With an incidence of 1 in 6,250 livebirths and a sibling recurrence risk of 3% (ref. 5), the risk ratio (λ) for S-HSCR is 187.5, which allowed us to identify the major segregating risk factors6. Consequently, we could both impute or refute the role of all known candidate genes (see Methods), independently assess the role of RET and identify new genes involved in HSCR. As previous segregation analysis of S-HSCR could not specify a precise genetic model, we used model-free linkage analyses, which make no assumptions about the numbers and properties of segregating factors11,12,13,14.
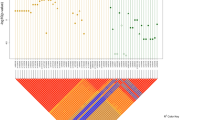
We carried out allele-sharing linkage analysis and detected significant evidence of co-segregation by the disease with markers on chromosome 10q11 between RETint5 and D10S1099 (Z=5.02, P=2.4 × 10−7) at the RET locus (Table 1 and Fig. 1). Two other regions showed some evidence of linkage: chromosome 3p21, between markers D3S2408 and D3S1766 (Z=3.46, P=1.9 × 10−4), and chromosome 19q12, between markers D19S1037 and D19S587 (Z=3.23, P=6.6 × 10−4). To validate these results, we carried out two additional tests. First, we showed that the gene on chromosome 10q11 conferring susceptibility is RET by conducting extensive mutation analysis of the RET coding sequence in all 49 families. We detected 17 sequence changes, relative to the wildtype sequence, excluding known polymorphisms: ten probands had missense changes, four probands had nonsense or frameshift mutations, and three families harbored sequence alterations that could affect normal splicing (Fig. 2). Thus, RET mutations probably explain all affected families showing linkage to 10q11, demonstrating the significant effect of RET in S-HSCR, although mutations in another closely linked gene cannot be ruled out. Segregation at 10q11 is not sufficient to explain the inheritance of S-HSCR, however, as the expected probability of sharing 0 alleles identical-by-descent (IBD) is 1/4λ=0.0013 (λ=187.5). Assuming a Poisson distribution, the probability of observing two or more sibling pairs with IBD=0 is 8.8 × 10−7.

Each panel, indicated by the chromosome number, shows the genetic length of the chromosome in cM on the x axis and the nonparametric linkage (NPL) score on the y axis. The red dots signify the locations of HSCR candidate genes. The red and green dashed lines represent statistical thresholds for significant and suggestive linkage.

RET mutations, when identified in the coding sequence and intron–exon boundaries, were classified as 'severe' (red) when the amino-acid change was nonsense or nonconservative or altered a residue evolutionarily conserved among RET sequences of mouse, chicken, zebrafish and Drosophila; otherwise, the change was termed 'mild' (blue). The indicated domains of RET are: SP, signal peptide; CAD, cadherin-like; CYS, cysteine-rich; TM, transmembrane segment; TK1, TK2, tyrosine kinase domains.
Second, we re-estimated the significance values of all significant and suggestive linkages by using the computer program GENEHUNTER+13. Only segregation of markers at 10q11, 3p21 and 19q12 are significant13 and mean allele-sharing at each of the three loci is 75%, 69% and 70%, respectively (Table 1). Many genes in the signaling cascades mediated by RET and endothelin receptor type B have been suggested as candidates underlying HSCR5. The lack of evidence of linkage at their genomic map locations suggests that none of these genes, except RET, contributes significantly to S-HSCR risk. This view is supported by previous mutation analysis of these same candidate genes5.
The magnitude of the genetic effect of each mapped locus on HSCR susceptibility can be estimated from the proportion of affected sibling pairs that show IBD=0 (ref. 15). Thus, the locus-specific risk ratio conferred by the 10q11, 3p21 and 19q12 factors is 8.3-, 4.2- and 5.0-fold, respectively (Table 1). In addition, the estimated IBD=0 probabilities, when compared with the null hypothesis value of 25%, provide an estimate of the proportion of families showing segregation at each mapped factor: 88%, 76% and 80% of families at the 10q11, 3p21 and 19q12 loci, respectively. The joint classification of sharing in the 67 affected sibling pairs fits a multiplicative model of genetic effects (Table 2). Under this model, the predicted phenotypic risk ratio for siblings of a proband carrying susceptibility alleles at all three loci is 8.3 × 4.2 × 5.0, or a 174.3-fold increase, which is similar to the previously observed value of 187.5 (ref. 6). A parsimonious hypothesis is that segregation of the three loci at 10q11, 3p21 and 19q12 is both necessary and sufficient to explain disease aggregation.
The linkage data provide additional clues regarding the mode of inheritance of S-HSCR. The frequency of sharing one allele IBD is approximately 50% for each locus (Table 1); thus, the transmission of susceptibility alleles follows an additive model. For rare mutations, inheritance is probably from a single parent or in a dominant manner (at least 1,000-fold more likely than recessive models). Under this assumption, we carried out parametric analyses to estimate the frequency of the allele conferring susceptibility. We maximized the lod score by varying allele frequency and penetrance (2:1 for male:female) across a grid (frequencies of 0.0001, 0.001 and 1% increments from 0.01 to 0.10; penetrance of 58/29, 80/40 and 40/20). We obtained maximum lod scores of 6.85, 3.58 and 3.26 for the chromosomes 10q11, 3p21 and 19q12 loci, respectively, at susceptibility-allele frequencies of 1%, 4% and 5%, respectively. As a consequence, and under the multiplicative model of penetrance, the frequency of individuals heterozygous with respect to all three loci, the most common at-risk genotype, is 14/100,000 and close to the observed population incidence (Table 2). The proposed genetic model is therefore consistent with population incidence.
We explored whether additional genetic models of multigenic inheritance are compatible with the data. We used a parametric variance components method15 to test whether different hypotheses about mode of inheritance fit familial patterns (Table 3). Three major conclusions emerged. First, among two-locus comparisons, chromosome 10 exerts the major effect (the likelihood of the segregation pattern is 1,000-fold or greater as compared with the chromosome 3 and 19 loci). Second, all three-locus models show better fit than any two-locus model. Among three-locus models, a simple heterogeneity (additive) model, where any single locus can lead to HSCR, provides adequate fit; however, the fit is improved by a million-fold when a multiplicative model is assumed. Alternatively, a model where S-HSCR arises from interactions between the 10q11 and 3p21 loci in some families and the 10q11 and 19q12 loci in the remaining families shows as good a fit as the multiplicative model. A model involving epistasis (nonadditive interaction) between all three loci also fits the data, but requires the estimation of an additional parameter. When each model is compared to the full model, the parsimonious conclusion is that the multiplicative mode of inheritance is the best hypothesis. Thus, on the basis of the three-locus segregation, prediction of disease incidence and familial risk, we conclude that additional major gene effects probably do not exist.
We showed conclusively that the susceptibility gene at chromosome 10q11 is RET. However, as the estimated proportion of RET-linked families was 88% (corresponding to 43 of 49 families), the identification of 17 mutations in 43 linked families (40%) is surprising. We suggest that noncoding mutations probably make a significant contribution to S-HSCR; such sequence changes may represent low-penetrance alleles16,17,18. As observed for the coding sequence mutations, the noncoding mutations are expected to be diverse or recurrent, as we found no allelic associations with RET polymorphisms. Recent studies of isolated HSCR have shown association with the RET polymorphisms Ala45Ala (exon 2) and Leu769Leu (exon 13)19,20. However, in our familial cases, frequencies of A45A and L769L are not different from controls (see Methods). The mutations show a nonrandom distribution within RET, with a majority (16/17 changes) of changes in the extracellular domains (Fig. 2). This contrasts with mutations identified in families with multigenerational HSCR, where sequence changes are observed throughout the gene5. The molecular basis for this difference, and how it might relate to S- versus L-HSCR, is unknown.
Previously described multiplex families show a greater than expected inheritance of HSCR through the maternal lineage. Of the 49 families studied here, 27 share one allele IBD at RET; although we expect the shared allele to be equally transmitted by either parent, we observed, instead, 21 maternal and 6 paternal transmissions (P<0.003; Table 1). This effect is not gender-specific but a true parent-of-origin effect, as, within the 27 nuclear families with IBD=1, there are 29 affected males and 25 affected females. Similar analysis of segregation at 3p21 and 19q12 showed no parent-of-origin effects (Table 1). Consequently, the transmission bias observed at the phenotypic level is associated with RET markers and not with those on chromosomes 3 and 19.
We recently reported evidence of a chromosome 9q31 locus that increases HSCR susceptibility in families with L-HSCR with 'mild' RET alleles (see Methods)7. No significant sharing was detected for this region in the families described here (Z=1.52, P=0.065), yet the location of a peak maps to approximately the same region (within 15 cM). In our previous study7, we detected significant results only when the nature of the RET mutation was considered. We reanalyzed the present data by classifying families according to the presence or absence of a 'severe' RET mutation: 9 families harbored a 'severe' mutation with random sharing at 9q31 (P=0.55), and the remaining 40 had 'mild' or undetected changes with nonrandom allele sharing (P=0.027). These are precisely the nature of the results observed in families with L-HSCR, suggesting the action of a modifier locus at 9q31 that is RET-dependent, although this effect is weaker in S-HSCR.
We have shown evidence of oligogenic inheritance in a complex human disease. The action of three loci, RET and two as-yet unidentified genes on chromosomes 3p21 and 19q12, seem to be necessary and sufficient for S-HSCR, although a model where S-HSCR arises from the action of either RET and a 3p21 gene or RET and a 19q21 gene cannot be currently refuted. These three genes seem to act in all families, as subdivision of families by the presence of L-HSCR versus S-HSCR or by coding versus noncoding mutations does not alter any of the conclusions. Our failure to detect segregation of all three loci in some families does not contradict the joint action of all three loci, as the presence of new mutations or homozygous susceptibility factors, in a collection of ascertained families, cannot be negated. The evidence that S-HSCR does not usually segregate in the absence of RET (Table 3) suggests that the actions of the chromosome 3p21 and 19q12 loci are RET-dependent, and are, consequently, modifiers of RET expression. Given the estimated high frequencies (4% and 5%) of the putative variants, these alleles as a class are polymorphic in the human population, although individual alleles may be rare. We have shown that noncoding mutations in RET are important in S-HSCR and that their frequency is probably higher in S- than in L-HSCR7. This latter factor may contribute to the lower penetrance in S-versus L-HSCR16,17,18. Finally, the three loci detected recapitulate all major aspects of the genetic epidemiology of S-HSCR, suggesting that this approach can be extended to dissect complex genetic diseases using contemporary genetic tools.
Methods
Families studied.
We ascertained 61 multiplex families after obtaining informed consent and classified them into those with three or more affected individuals in several generations (group A enriched for L-HSCR: 12 families), and multiplex nuclear families with no further evidence of HSCR (group B enriched for S-HSCR: 49 families). The affected sibships (group B) had 2, 3 or 4 affected individuals in 43, 4 and 2 families, respectively, comprising 67 distinct affected sibling pairs and 106 affected individuals. The parents in 46 families were clinically unaffected; the remaining families had one affected parent (2 male, 1 female) and two affected children each. The affected individuals were enriched for males (1.54:1) and S-HSCR (40/62). We extracted genomic DNA and established lymphoblastoid cell lines from participating members using standard techniques.
Genome scan markers.
We carried out a genome scan using microsatellite markers from the Weber screening set 8.0 and additional markers from Genethon. We reconstructed the meiotic map based on markers used and MultiMap (see Web Table A online)21. This genetic map spanned 3,669 cM and covered over 92% of the human genome. We carried out genotyping using semi-automated fluorescence-based protocols on an ABI 377 automated sequencer (Perkin-Elmer). We assembled PCRs in 96-well formats using a multichannel pipettor to aliquot 1 μl of genomic DNA (20 ng) and a Hamilton workstation to dispense 4 μl of a PCR mixture (10 mM Tris-HCl, 50 mM KCl, 0.75–3.0 mM MgCl2, 200 mM dNTPs, 0.3 mM each of dye-labeled forward and unlabeled reverse primer and 0.125 U Taq polymerase), for a final volume of 5 μl. We carried out 31 cycles (15 s at 94 °C, 10 s at 55–58 °C, 15 s at 72 °C) of PCR using MJR Tetrad thermocyclers. Approximately 1.5 μl of each of 10–12 PCRs for each DNA sample were pooled using a Hamilton workstation, of which 0.7 μl was combined with loading solution (deionized formamide, 50 μg μl−1 Dextran Blue) and a TAMRA-500 molecular weight size standard, heat-denatured and loaded onto the ABI 377 for gel electrophoresis. We assigned allele sizes using Genescan v. 2.02 and Genotyper v. 1.0 software and using the known genotype of a reference individual (CEPH 1331-01).
Linkage analysis.
Allele frequencies were estimated from 86 of the 98 unrelated parents and compared with those in public databases; no statistically significant differences were identified. All alleles were recoded into integers, and mendelian inheritance was assessed within families using PedCheck22; we re-analyzed or re-genotyped all inconsistencies to correct errors. We used the genotypes for all 371 markers and the 'kinship' program within ASPEX to verify relationships between individuals in each family. This likelihood analysis verified the full sibling relationships among all affected individuals in each nuclear family. We used GENEHUNTER11,12 to estimate the genetic length of each chromosome in each family to check for gross genotyping errors resulting in map inflation; we found no significant differences. To assess genotyping accuracy, we re-genotyped 22 markers in 96 DNA samples and detected 5 discrepancies, for an error rate of approximately 0.2%.
Genes relevant to HSCR specifically tested for linkage include: (i) RET, GDNF, EDNRB, EDN3 and SOX10, given their known roles in HSCR pathology5; (ii) additional components of RET signaling: NRTN, neurturin, an alternative RET ligand23; GFRA1, a GPI-linked RET co-receptor24,25 and GRB10, a RET adaptor protein26; (iii) an additional component of endothelin receptor type B signaling, ECE1, an EDN3 processing enzyme27; (iv) a knockout mouse model of aganglionosis and craniofacial abnormalities, DLX2 (ref. 28) and (v) IKBKAP, a gene underlying familial dysautonomia29.
RET mutation analysis.
We carried out mutation analysis by screening 21 exons of RET comprising 3,474 bp and 1,700 bp of adjacent intronic sequence as wildtype standards. We screened one affected individual from each family and used three methods on each sample: single-strand conformational polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE) and automated nucleotide sequencing using dye terminator chemistry. A sequence alteration was defined as a mutation if it failed to occur in a group of 100 control individuals; otherwise, it was termed a variant or polymorphism. All mutations were classified by the nature of the predicted change in the RET protein as 'severe' if the substitution was either a nonconservative change based on BLOSUM62 (ref. 30) or any change at an evolutionary conserved site (defined as a site unchanged in protein sequence comparisons of human, mouse, Drosophila, zebrafish and C. elegans RET), or 'mild' if the change was conservative.
Test of allelic association between RET and HSCR.
We genotyped five common RET polymorphisms (Ala45Ala: exon 2, Ala432Ala: exon 7, Gly691Ser: exon 11, Leu769Leu: exon 13 and Ser904Ser: exon 15) in 47 probands and found no significant allele frequency differences between the 15 probands with an identified mutation (47%, 53%, 33%, 33%, 27%, respectively) and the 22 probands without an identified mutation (53%, 47%, 22%, 31%, 16%, respectively), and, indeed, all probands, with reported population frequencies19,20.
URLs.
Marker characteristics and allele frequencies were obtained from the public databases http://www.chlc.org, http://www.genethon.fr, http://www.resgen.com and http://www.marshmed.org (Weber screening set 8.0). The computer program ASPEX was obtained from http://www.lahmed.stanford.edu/pub/aspex.
GenBank accession numbers.
RET genomic data, AJ243297; RET mRNA, X15262.
Note: Supplementary information is available on the Nature Genetics website.
References
Chakravarti, A. Population genetics—making sense out of sequence. Nature Genet. 21, 56–60 (1999).
Lander, E.S. & Schork, N.J. Genetic dissection of complex traits. Science 265, 2037–2048 (1994).
Lander, E.S. The new genomics: global views of biology. Science 274, 536–539 (1996).
Holschneider, A.M. Hirschsprung's Disease (Thieme-Stratton, New York, 1982).
Chakravarti, A. & Lyonnet, S. Hirschsprung disease. in The Metabolic and Molecular Bases of Inherited Disease 8th edn (eds Scriver, C.R. et al.) 6231–6255 (McGraw Hill, New York, 2001).
Badner, J.A., Sieber, W.K., Garver, K.L. & Chakravarti, A. A genetic study of Hirschsprung disease. Am. J. Hum. Genet. 46, 568–580 (1990).
Bolk, S. et al. A human model for multigenic inheritance: phenotypic expression in Hirschsprung disease requires both the RET gene and a new 9q31 locus. Proc. Natl Acad. Sci. USA 97, 268–273 (2000).
Attié, T. et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum. Mol. Genet. 4, 1381–1386 (1995).
Puffenberger, E.G. et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 79, 1257–1266 (1994).
Schuchardt, V., D'Agati, L., Larsson-Blomberg, L., Costantini, F. & Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367, 380–383 (1994).
Kruglyak, L., Daly, M.J., Reeve-Daly, M.P. & Lander, E.S. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 58, 1347–1363 (1996).
Kruglyak, L. & Lander, E.S. Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am. J. Hum. Genet. 57, 439–454 (1995).
Kong, A. & Cox, N.J. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 61, 1179–1188 (1997).
Lander, E.S. & Kruglyak, L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genet. 11, 241–247 (1995).
Olson, J.M. Likelihood-based models for genetic linkage analysis using affected sib pairs. Hum. Hered. 47, 110–120 (1997).
Sakai, T., Ohtani, N., McGee, T.L., Robbins, P.D. & Dryja, T.P. Oncogenic germ-line mutations on Sp1 and ATF sites in the human retinoblastoma gene. Nature 353, 83–86 (1991).
Otterson, G.A., Chen, W., Coxon, A.B., Khleif, S.N. & Kaye, F.J. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc. Natl Acad. Sci. USA 94, 12036–12040 (1997).
Rarve-Harel, N. et al. The molecular basis of partial penetrance of splicing mutations in cystic fibrosis. Am. J. Hum. Genet. 60, 87–94 (1997).
Fitze, G. et al. Association of RET protooncogene codon 45 polymorphism with Hirschsprung disease. Am. J. Hum. Genet. 65, 1469–1473 (1999).
Borrego, S. et al. RET genotypes comprising specific haplotypes of polymorphic variants predispose to isolated Hirschsprung disease. J. Med. Genet. 37, 572–578 (2000).
Matise, T.C. et al. An automated construction of genetic linkage maps using an expert system (MultiMap): a human genome linkage map. Nature Genet. 6, 384–390 (1994).
O'Connell, J.R. & Weeks, D.E. PedCheck: a program for identification of genotyping compatibilities in linkage analysis. Am. J. Hum. Genet. 63, 259–266 (1998).
Doray, B. et al. Mutation of the RET ligand, neurturin, supports multigenic inheritance in Hirschsprung disease. Hum. Mol. Genet. 7, 1449–1452 (1998).
Angrist, M. et al. Human GFRA1: cloning, mapping, genomic structure, and evaluation as a candidate gene for Hirschsprung disease susceptibility. Genomics 48, 354–362 (1998).
Myers, S.M. et al. Investigation of germline GFR α-1 mutations in Hirschsprung disease. J. Med. Genet. 36, 217–220 (1999).
Angrist, M. et al. Genomic structure of the gene for the SH2 and pleckstrin homology domain-containing protein GRB10 and evaluation of its role in Hirschsprung disease. Oncogene 17, 3065–3070 (1998).
Hofstra, R.M. et al. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with Hirschsprung disease, cardiac defects, and autonomic dysfunction. Am. J. Hum. Genet. 64, 304–308 (1999).
Qiu M, et al. Null mutation of Dlx-2 results in abnormal morphogenesis of proximal first and second branchial arch derivatives and abnormal differentiation in the forebrain. Genes Dev. 9, 2523–2538 (1995).
Slaugenhaupt, S.A. et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 68, 598–605 (2001).
Henikoff, S. & Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA 89, 10915–10919 (1992).
Acknowledgements
We are indebted to members of the families with HSCR for sharing information and donating tissue samples; without them, and the continued assistance of the American Pseudo-obstruction & Hirschsprung Disease Society, this work would not be possible. We gratefully acknowledge the efforts of J. Scott, P. Edery, B. Doray, the French HSCR Consortium and the Association Française de la Maladie de Hirschsprung for family studies, and K. Bentley, C. Kashuk and S. Osmani for excellent technical assistance. This work was supported by funds from Case Western Reserve University, University Hospitals of Cleveland, National Institutes of Health, Association Française contre les Myopathies, INSERM and the Fondation pour la Recherche Médicale.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Gabriel, S., Salomon, R., Pelet, A. et al. Segregation at three loci explains familial and population risk in Hirschsprung disease. Nat Genet 31, 89–93 (2002). https://doi.org/10.1038/ng868
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng868
This article is cited by
-
Copy number variations in candidate genomic regions confirm genetic heterogeneity and parental bias in Hirschsprung disease
Orphanet Journal of Rare Diseases (2019)
-
Closing the ‘phenotype gap’ in precision medicine: improving what we measure to understand complex disease mechanisms
Mammalian Genome (2019)
-
Hirschsprung disease — integrating basic science and clinical medicine to improve outcomes
Nature Reviews Gastroenterology & Hepatology (2018)
-
Hirschsprung’s disease: clinical dysmorphology, genes, micro-RNAs, and future perspectives
Pediatric Research (2017)
-
Exome-Wide Association Study Identified New Risk Loci for Hirschsprung’s Disease
Molecular Neurobiology (2017)
