
Abstract
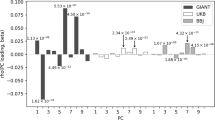
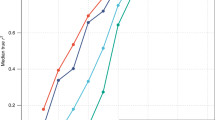
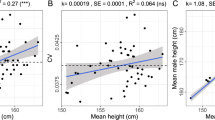
Strong signatures of positive selection at newly arising genetic variants are well documented in humans1,2,3,4,5,6,7,8, but this form of selection may not be widespread in recent human evolution9. Because many human traits are highly polygenic and partly determined by common, ancient genetic variation, an alternative model for rapid genetic adaptation has been proposed: weak selection acting on many pre-existing (standing) genetic variants, or polygenic adaptation10,11,12. By studying height, a classic polygenic trait, we demonstrate the first human signature of widespread selection on standing variation. We show that frequencies of alleles associated with increased height, both at known loci and genome wide, are systematically elevated in Northern Europeans compared with Southern Europeans (P < 4.3 × 10−4). This pattern mirrors intra-European height differences and is not confounded by ancestry or other ascertainment biases. The systematic frequency differences are consistent with the presence of widespread weak selection (selection coefficients ∼10−3–10−5 per allele) rather than genetic drift alone (P < 10−15).
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Tishkoff, S.A. et al. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science 293, 455–462 (2001).
Hamblin, M.T. & Di Rienzo, A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am. J. Hum. Genet. 66, 1669–1679 (2000).
Bersaglieri, T. et al. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 74, 1111–1120 (2004).
The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861 (2007).
Sabeti, P.C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007).
Voight, B.F., Kudaravalli, S., Wen, X. & Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006).
Williamson, S.H. et al. Localizing recent adaptive evolution in the human genome. PLoS Genet. 3, e90 (2007).
Hancock, A.M. et al. Adaptations to climate-mediated selective pressures in humans. PLoS Genet. 7, e1001375 (2011).
Hernandez, R.D. et al. Classic selective sweeps were rare in recent human evolution. Science 331, 920–924 (2011).
Pritchard, J.K. & Di Rienzo, A. Adaptation—not by sweeps alone. Nat. Rev. Genet. 11, 665–667 (2010).
Novembre, J. & Di Rienzo, A. Spatial patterns of variation due to natural selection in humans. Nat. Rev. Genet. 10, 745–755 (2009).
Hermisson, J. & Pennings, P.S. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169, 2335–2352 (2005).
Sabeti, P.C. et al. Positive natural selection in the human lineage. Science 312, 1614–1620 (2006).
Przeworski, M., Coop, G. & Wall, J.D. The signature of positive selection on standing genetic variation. Evolution 59, 2312–2323 (2005).
Barrett, R.D. & Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008).
Pritchard, J.K., Pickrell, J.K. & Coop, G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr. Biol. 20, R208–R215 (2010).
Orr, H.A. Testing natural selection versus genetic drift in phenotypic evolution using quantitative trait locus data. Genetics 149, 2099–2104 (1998).
Lewontin, R.C. Race and intelligence. Bull. At. Sci. 26, 2–8 (1970).
Cavelaars, A.E. et al. Persistent variations in average height between countries and between socio-economic groups: an overview of 10 European countries. Ann. Hum. Biol. 27, 407–421 (2000).
Lango Allen, H. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838 (2010).
Myocardial Infarction Genetics Consortium. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 41, 334–341 (2009).
Nelson, M.R. et al. The Population Reference Sample, POPRES: a resource for population, disease, and pharmacological genetics research. Am. J. Hum. Genet. 83, 347–358 (2008).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Campbell, C.D. et al. Demonstrating stratification in a European American population. Nat. Genet. 37, 868–872 (2005).
Hirschhorn, J.N. & Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108 (2005).
Freedman, M.L. et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 36, 388–393 (2004).
Lander, E.S. & Schork, N.J. Genetic dissection of complex traits. Science 265, 2037–2048 (1994).
Ayodo, G. et al. Combining evidence of natural selection with association analysis increases power to detect malaria-resistance variants. Am. J. Hum. Genet. 81, 234–242 (2007).
The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Simonson, T.S. et al. Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 (2010).
Splansky, G.L. et al. The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol. 165, 1328–1335 (2007).
Acknowledgements
The authors would like to thank E.L. Altmaier, K.E. Samocha, S.R. Grossman, G. Coop, other attendees of the Biology of Genomes 2011 conference, B.F. Voight, M. McCarthy, P. Visscher and other members of the Reich and Hirschhorn labs for their discussions and helpful comments. We gratefully thank the GIANT consortium and particularly the members of the height working group for making unpublished association data available. We thank the MIGen consortium for making allele frequency data available. This research was conducted using data and resources from the FHS of the National Heart, Lung, and Blood Institute of the US National Institutes of Health and Boston University School of Medicine based on analyses by FHS investigators participating in the SNP Health Association Resource project. This work was supported by the National Heart, Lung and Blood Institute's FHS (contract no. N01-HC-25195) and its contract with Affymetrix, Inc., for genotyping services (contract no. N02-HL-6-4278). A portion of this research used the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center. This work was also supported by a graduate research fellowship from the National Science Foundation (to C.W.K.C.), the March of Dimes (6-FY09-507 to J.N.H.) and the National Institute of Diabetes and Digestive and Kidney Diseases (1R01DK075787 to J.N.H.).
Author information
Authors and Affiliations
Consortia
Contributions
M.C.T., C.W.K.C., C.D.P., S.S., D.R. and J.N.H. conceived of and designed the experiments; M.C.T. and C.D.P. performed the analyses; M.C.T., C.W.K.C. and J.N.H. interpreted the data; C.W.K.C., C.D.P., D.R. and the GIANT Consortium contributed materials; M.C.T., C.W.K.C. and J.N.H. wrote the paper with input from all coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
A full list of members and institutions is provided in the Supplementary Note.
Rights and permissions
About this article
Cite this article
Turchin, M., Chiang, C., Palmer, C. et al. Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nat Genet 44, 1015–1019 (2012). https://doi.org/10.1038/ng.2368
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.2368
This article is cited by
-
Is there still evolution in the human population?
Biologia Futura (2022)
-
Genomic approaches to trace the history of human brain evolution with an emerging opportunity for transposon profiling of ancient humans
Mobile DNA (2021)
-
Multi-omic profiling of pituitary thyrotropic cells and progenitors
BMC Biology (2021)
-
Selection on adaptive and maladaptive gene expression plasticity during thermal adaptation to urban heat islands
Nature Communications (2021)
-
Admixture-enabled selection for rapid adaptive evolution in the Americas
Genome Biology (2020)
