
Abstract
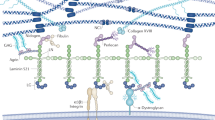
The glomerular basement membrane (GBM) is a specialized form of basement membrane that has a major role in the maintenance of the glomerular filtration barrier. Like all basement membranes, it contains four main components: type IV collagen, laminin, nidogen, and heparan sulfate proteoglycans. Different isoforms of these large molecules are produced. These isoforms have a tissue-specific distribution; in the mature GBM, the major type IV collagen molecule is the α3α4α5(IV) isoform, associated with laminin-521 (α5β2γ1), nidogen and agrin heparan sulfate proteoglycans. The importance of the GBM has been demonstrated by identification of hereditary glomerular diseases linked to structural anomalies of its components; for example, type IV collagen in Alport syndrome and familial benign hematuria, and laminin in Pierson syndrome. Type III collagen, an interstitial collagen, accumulates within the GBM of patients with the nail–patella syndrome, and abnormal deposition of fibronectin, another extracellular matrix protein, is characteristic of so-called fibronectin nephropathy. Development of animal models of these diseases has facilitated precise analysis of pathogenic mechanisms, but no specific treatments are available. Therapeutic trials in Alport syndrome nephropathy are underway, following promising preliminary results obtained in rodent and canine models of the disorder.
Key Points
-
No specific treatments are available for inherited diseases in which mutation of genes that encode components of the glomerular basement membrane (GBM) perturb its structure
-
Hematuria is a major clinical feature of Alport syndrome, a progressive disease in which the structure of type IV collagen in the GBM is abnormal
-
Alport syndrome can be inherited in an X-linked dominant, autosomal recessive or autosomal dominant manner
-
Laminin glycoproteins are essential to the assembly of the GBM and mutations of LAMB2, which encodes the β2 chain of laminin, lead to Pierson syndrome
-
Deposition of type III collagen within the GBM is the hallmark of nail–patella syndrome and accumulation of this protein in the glomerular extracellular matrix is also observed in rare nonsyndromic glomerulopathies
-
Parietal and mesangial deposition of fibronectin also cause glomerulopathy
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Timpl R (1989) Structure and biochemical activity of basement membrane proteins. Eur J Biochem 180: 487–502
Hudson BG et al. (2003) Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med 348: 2543–2556
Soininen R et al. (1988) The structural genes for α1 and α2 chains of human type IV collagen are divergently encoded on opposite DNA strands and have an overlapping promoter region. J Biol Chem 263: 17217–17220
Mariyama M et al. (1992) Colocalization of the genes for the α3(IV) and α4(IV) chains of type IV collagen to chromosome 2 bands q35–q37. Genomics 13: 809–813
Leinonen A et al. (1994) Complete primary structure of the human type IV collagen α4(IV) chain: comparison with structure and expression of the other α(IV) chains. J Biol Chem 269: 26172–26177
Mariyama M et al. (1994) Complete primary structure of the human α3(IV) collagen chain: coexpression of the α3(IV) and α4(IV) collagen chains in human tissues. J Biol Chem 269: 23013–23017
Hostikka SL et al. (1990) Identification of a distinct type IV collagen α chain with restricted kidney distribution and assignment of its gene to the locus of X-linked Alport syndrome. Proc Natl Acad Sci USA 87: 1606–1610
Myers JC et al. (1990) Molecular cloning of α5(IV) collagen and assignment of the gene to the region of the X chromosome containing the Alport syndrome locus. Am J Hum Genet 46: 1024–1033
Sugimoto M et al. (1994) The genes COL4A5 and COL4A6, coding for basement membrane collagen chains α5(IV) and α6(IV), are located head-to-head in close proximity on human chromosome Xq22 and COL4A6 is transcribed from two alternative promoters. Proc Natl Acad Sci USA 91: 11679–11683
Miner JH and Sanes JR (1994) Collagen IV α3, α4, and α5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J Cell Biol 127: 879–891
Kalluri R et al. (1997) Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J Clin Invest 99: 2470–2478
Cheong HI et al. (1994) Immunohistologic studies of type IV collagen in anterior lens capsules of patients with Alport syndrome. Lab Invest 70: 553–557
Cosgrove D et al. (1996) Immunohistochemical localization of basement membrane collagens and associated proteins in the murine cochlea. Hear Res 97: 54–65
Borza DB et al. (2001) The NC1 domain of collagen IV encodes a novel network composed of the α1, α2, α5, and α6 chains in smooth muscle basement membranes. J Biol Chem 276: 28532–28540
Peissel B et al. (1995) Comparative distribution of the α1(IVα5(IV), and α6(IV) collagen chains in normal human adult and fetal tissues and in kidneys from X-linked Alport syndrome patients. J Clin Invest 96: 1948–1957
Ninomiya Y et al. (1994) Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J Cell Biol 130: 1219–1229
Yurchenco PD and Ruben GC (1987) Basement membrane structure in situ: evidence for lateral associations in the type IV collagen network. J Cell Biol 105: 2559–2568
Timpl R and Brown JC (1994) The laminins. Matrix Biology 14: 275–281
Miner JH (1999) Renal basement membrane components. Kidney Int 56: 2016–2024
Aumailley M et al. (2005) A simplified laminin nomenclature. Matrix Biol 24: 326–332
Katz A et al. (1991) Renal entactin (nidogen): isolation, characterization and tissue distribution. Kidney Int 10: 643–652
Groffen AJ et al. (1998) Agrin is a major heparan sulfate proteoglycan in the human glomerular basement membrane. J Histoch Cytoch 46: 19–27
Alport AC (1927) Hereditary familial congenital haemorrhagic nephritis. Br Med J 1: 504–506
Atkin CL et al. (1988) Alport syndrome. In Diseases of the Kidney, edn 4, 617–641 (Eds Schrier RW and Gottschalk CW) Boston: Little, Brown, and Company
Flinter FA et al. (1988) Genetic of classic Alport's syndrome. Lancet 2: 1005–1007
Gubler M et al. (1981) Alport's syndrome: a report of 58 cases and a review of the literature. Am J Med 70: 493–505
Kashtan CE and Michael AF (1993) Alport syndrome: from bedside to genome to bedside. Am J Kidney Dis 22: 627–640
Pajari H et al. (1996) Alport's syndrome in 78 patients: epidemiological and clinical study. Acta Paediatr 85: 1300–1306
Nielsen CE (1977) Lenticonus anterior and Alport's syndrome. Acta Ophthalmol 56: 518–530
Polack BCP and Hogewind BL (1977) Macular lesions in Alport's disease. Am J Ophthalmol 84: 533–535
Perrin D et al. (1980) Perimacular changes in Alport's syndrome. Clin Nephrol 13: 163–167
Rhys C et al. (1997) Recurrent corneal erosion associated with Alport's syndrome. Kidney Int 52: 208–211
Hinglais N et al. (1972) Characteristic ultrastructural lesion of the glomerular basement membrane in progressive hereditary nephritis (Alport's syndrome). Lab Invest 27: 473–487
Spear GS and Slusser RJ (1972) Alport's syndrome: emphasizing electron microscopic studies of the glomerulus. Am J Pathol 69: 213–222
Churg J and Sherman RL (1973) Pathologic characteristics of hereditary nephritis. Arch Pathol 95: 374–379
Gubler MC et al. (2006) Alport syndrome, familial benign hematuria, nail–patella syndrome and type III collagen nephropathy. In Heptinstall's Pathology of the Kidney, edn 6, 487–515 (Eds Jennette JC et al.) Baltimore: Lippincott, Williams & Wilkins
Jais JP et al. (2000) X-linked Alport syndrome: natural history in 195 families and genotype–phenotype correlations in males. J Am Soc Nephrol 11: 649–657
Barker DF et al. (1990) Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248: 1224–1227
Mochizuki T et al. (1994) Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8: 77–82
Gross O et al. (2002) Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: impact on clinical counseling. Nephrol Dial Transplant 17: 1218–1227
Jais JP et al. (2003) X-Linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J Am Soc Nephrol 14: 2603–2610
Tryggvason K et al. (1993) Molecular genetics of Alport syndrome. Kidney Int 43: 38–44
Lemmink HH et al. (1997) The clinical spectrum of type IV collagen mutations. Hum Mutat 9: 477–499
Antignac C et al. (1994) Deletions in the COL4A5 collagen gene in X-linked Alport syndrome: characterization of the pathological transcripts in nonrenal cells and correlation with diseases expression. J Clin Invest 93: 1195–1207
Renieri A et al. (1995) Major COL4A5 gene rearrangements in patients with juvenile type Alport syndrome. Am J Med Genet 59: 380–385
Knebelmann B et al. (1996) Spectrum of mutations in the COL4A5 gene in X-linked Alport syndrome. Am J Hum Genet 59: 1221–1232
Kawai S et al. (1996) The COL4A5 gene in Japanese Alport syndrome patients: spectrum of mutations of all exons. Kidney Int 49: 814–822
Martin P et al. (1998) High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J Am Soc Nephrol 9: 2291–2301
Inoue Y et al. (1999) Detection of mutations in the COL4A5 gene in over 90% of male patients with X-linked Alport's syndrome by RT-PCR and direct sequencing. Am J Kidney Dis 34: 854–862
Barker DF et al. (1996) A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am J Hum Genet 58: 1157–1165
Antignac C et al. (1992) Alport syndrome and diffuse leiomyomatosis: deletions in the 5′ end of the COL4A5 collagen gene. Kidney Int 42: 1178–1183
Zhou J et al. (1993) Deletion of the paired α5(IV) and α6(IV) collagen genes in inherited smooth muscle tumors. Science 261: 1167–1169
Heidet L et al. (1995) Deletions of both α5(IV) and α6(IV) collagen genes in Alport syndrome and in smooth muscle cell proliferation associated with Alport syndrome. Hum Mol Genet 4: 99–108
Vitelli F et al. (1999) Identification and characterization of a highly conserved protein absent in the Alport syndrome (A), mental retardation (M), midface hypoplasia (M), and elliptocytosis (E) contiguous gene deletion syndrome (AMME). Genomics 55: 335–340
Nakanishi K et al. (1994) Immunohistochemical study of α1–5 chains of type IV collagen in hereditary nephritis. Kidney Int 46: 1413–1421
Mazzucco G et al. (1998) Ultrastructural and immunohistochemical findings in Alport's syndrome: a study of 108 patients from 97 Italian families with particular emphasis on COL4A5 gene mutation correlations. J Am Soc Nephrol 9: 1023–1031
Kashtan C and Kim Y (1992) Distribution of the α1 and α2 chains of collagen IV and of collagens V and VI in Alport syndrome. Kidney Int 42: 115–126
Lemmink HH et al. (1994) Mutations in the type IV collagen α3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet 3: 1269–1273
Boye E et al. (1998) Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am J Hum Genet 63: 1329–1340
Heidet L et al. (2001) Structure of the human type IV collagen gene COL4A3 and mutations in autosomal Alport syndrome. J Am Soc Nephrol 12: 97–106
Longo I et al. (2006) Autosomal recessive Alport syndrome: an in-depth clinical and molecular analysis of five families. Nephrol Dial Transplant 21: 665–671
Gubler MC et al. (1995) Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int 47: 1142–1147
Rogers PW et al. (1973) Familial benign essential hematuria. Arch Intern Med 131: 257–262
Van der Loop FT et al. (2000) Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney Int 58: 1870–1875
Pescucci C et al. (2004) Autosomal-dominant Alport syndrome: natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int 65: 1598–1603
Savige J et al. (2003) Thin basement membrane nephropathy. Kidney Int 64: 1169–1178
Tryggvason K and Patrakka J (2006) Thin basement membrane nephropathy. J Am Soc Nephrol 17: 813–822
Kashtan CE (1998) Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol 9: 1736–1750
Lemmink HH et al. (1996) Benign familial hematuria due to mutation of the type IV collagen α4 chain. J Clin Invest 98: 1114–1118
Buzza M et al. (2001) Segregation of hematuria in thin basement membrane disease with haplotypes at loci for Alport syndrome. Kidney Int 59: 1670–1675
Badenas C et al. (2002) Mutations in the COL4A4 and COL4A3 genes cause familial benign hematuria. J Am Soc Nephrol 13: 1248–1254
Piccini M et al. (1999) Evidence for genetic heterogeneity in benign familial hematuria. Am J Nephrol 19: 464–467
Longo I et al. (2002) From benign familial hematuria to autosomal dominant or recessive Alport syndrome. Kidney Int 61: 1947–1956
Torra R et al. (2004) Collagen type IV (α3–α4) nephropathy from isolated haematuria to renal failure. Nephrol Dial Transplant 19: 2429–2432
Seri M et al. (2000) Mutations in MYH9 result in the May–Hegglin anomaly, and Fechtner and Sebastian syndromes: the May–Hegglin/Flechtner Syndrome Consortium. Nat Genet 26: 103–105
Tazón-Vega B et al. (2007) Genetic testing for X-linked Alport syndrome by direct sequencing of COL4A5 cDNA from hair root RNA samples. Am J Kidney Dis 50: 257.e1–257.e14
Heidet L et al. (2003) A human-mouse chimera of the α3α4α5(IV) collagen protomer rescues the renal phenotype in Col4a3−/− mice. Am J Pathol 163: 1633–1644
Sugimoto H et al. (2006) Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci USA 103: 7321–7326
Prodromidi EI et al. (2006) Bone marrow derived-cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells 24: 2448–2455
Gross O et al. (2004) Antifibrotic, nephroprotective potential of ACE inhibitor vs AT1 antagonist in a murine model of renal fibrosis. Nephrol Dial Transplant 67: 1716–1723
Ninichuk V et al. (2005) Delayed chemokine receptor 1 blockade prolongs survival in collagen 4A3-deficient mice with Alport disease. J Am Soc Nephrol 16: 977–985
Koepke Ml et al. (2007) Nephroprotective effect of the HMG-CoA-reductase inhibitor cerivastatin in a mouse model of progressive renal fibrosis in Alport syndrome. Nephrol Dial Transplant 22: 1062–1069
Zeisberg M et al. (2006) Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PloS Med 3: e100
Zhang KW et al. (2007) Do mutations in COL4A1 or COL4A2 cause thin basement membrane nephropathy (TBMN)? Pediatr Nephrol 22: 645–651
Gould DB et al. (2005) Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308: 1167–1171
Van der Knaap MS et al. (2006) Neonatal porencephaly and adult stroke related to mutations in collagen IV A1. Ann Neurol 59: 504–511
Plaisier E et al. Role of COL4A1 mutations in the hereditary angiopathy with nephropathy, aneurysm and cramps (HANAC) syndrome. N Engl J Med, in press
Pierson M et al. (1963) Une curieuse association malformative congénitale et familiale atteignant l'oeil et le rein. J Génét Hum 12: 184–213
Zenker M et al. (2004) Human laminin β2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities (Pierson syndrome). Hum Mol Genet 13: 2625–2632
Hasselbacher K et al. (2006) Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int 70: 1008–1012
Noakes PG et al. (1995) The renal glomerulus of mice lacking s-laminin/laminin β2: nephrosis despite molecular compensation by laminin β1. Nat Genet 10: 400–406
Jarad G et al. (2006) Proteinuria precedes podocyte abnormalities in Lamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest 16: 2272–2279
Sweeney E et al. (2003) Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet 40: 153–162
Bongers EM et al. (2005) Genotype-phenotype studies in nail–patella syndrome show that LMX1B mutation location is involved in the risk of developing nephropathy. Eur J Hum Genet 13: 1019–1024
Heidet L et al. (2003) In vivo expression of putative LMX1B targets in nail–patella syndrome kidneys. Am J Pathol 163: 145–155
Ben Bassat M et al. (1971) The glomerular basement membrane in the nail–patella syndrome. Arch Pathol 92: 350–355
Hoyer JR et al. (1972) Renal disease in nail–patella syndrome: clinical and morphologic studies. Kidney Int 2: 231–238
Bennet WM et al. (1973) The nephropathy of the nail–patella syndrome: clinicopathologic analysis of 11 kindreds. Am J Med 54: 304–319
Vollrath D et al. (1998) Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail patella syndrome. Hum Mol Genet 7: 1091–1098
Chen H et al. (1998) Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet 19: 51–55
Dreyer SD et al. (1998) Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nat Genet 19: 47–50
Rohr C et al. (2002) The LIM-homeodomain transcription factor Lmx1b plays a crucial role in podocytes. J Clin Invest 109: 1073–1082
Miner JH et al. (2002) Transcriptional induction of slit diaphragm genes by Lmx1b is required in podocyte differentiation. J Clin Invest 109: 1065–1072
Morello R et al. (2001) Regulation of glomerular basement membrane collagen expression by LMX1B contributes to renal disease in nail patella syndrome. Nat Genet 27: 205–208
Salcedo JR (1984) An autosomal recessive disorder with glomerular basement membrane abnormalities similar to those seen in the nail–patella syndrome: report of a kindred. Am J Med Genet 19: 579–584
Arakawa M and Yamanaka N (1991) Collagenofibrotic glomerulonephropathy. London: Nishimura & Smith-Gordon
Gubler MC et al. (1993) Collagen type III glomerulopathy: a new type of hereditary nephropathy. Pediatr Nephrol 7: 354–360
Imbasciati E et al. (1991) Collagen type III glomerulopathy: a new idiopathic glomerular disease. Am J Nephrol 11: 422–429
Vogt BE et al. (1995) Inherited factor H deficiency and collagen type III glomerulopathy. Pediatr Nephrol 9: 11–15
Strom EH et al. (1995) Glomerulopathy associated with predo minant fibronectin deposits: a newly recognized hereditary disease. Kidney Int 48: 163–170
Assmann KJM et al. (1995) Familial glomerulonephritis characterized by massive deposits of fibronectin. Am J Kidney Dis 25: 781–791
Hildebrandt F et al. (1996) Glomerulopathy associated with predominant fibronectin deposits: exclusion of the genes for fibronectin, villin, and desmin. Am J Med Genet 63: 323–327
Zhang Z et al. (1997) Severe fibronectin-deposit renal glomerular disease in mice lacking uteroglobin. Science 276: 1408–1412
Volmer M et al. (1998) Exclusion of the uteroglobin gene as a candidate for fibronectin glomerulopathy (GFND). Nephrol Dial Transplant 13: 2417–2418
Vollmer M et al. (2000) Molecular cloning of the critical region for glomerulopathy with fibronectin deposits and evaluation of candidated genes. Genomics 68: 127–135
Acknowledgements
This work was supported by the Institut National de la Santé et de la Recherche Médicale and the Centre de référence Maladies Rénales Héréditaires de l'Enfant et de l'Adulte and by grants from the Association pour l'Utilisation du Rein Artificiel and the Association pour l'Information et la Recherche sur les Maladies Rénales Génétiques. I also thank E Le Gall for excellent technical assistance with figure preparation. Désirée Lie, University of California, Irvine, CA, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the Medscape-accredited continuing medical education activity associated with this article.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Gubler, M. Inherited diseases of the glomerular basement membrane. Nat Rev Nephrol 4, 24–37 (2008). https://doi.org/10.1038/ncpneph0671
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ncpneph0671
This article is cited by
-
Strong mesangial IgA staining—does it always refer to IgA nephropathy in a patient with proteinuria and hematuria? Answers
Pediatric Nephrology (2021)
-
Basement membranes in the cornea and other organs that commonly develop fibrosis
Cell and Tissue Research (2018)
