
Abstract
The asymmetric synthesis of N-allylic indoles is important for natural product synthesis and pharmaceutical research. The regio- and enantioselective N-allylation of indoles is a true challenge due to the favourable C3-allylation. We develop here a new strategy to the asymmetric synthesis of N-allylic indoles via rhodium-catalysed N-selective coupling of aryl hydrazines with allenes followed by Fischer indolization. The exclusive N-selectivities and good to excellent enantioselectivities are achieved applying a rhodium(I)/DTBM-Segphos or rhodium(I)/DTBM-Binap catalyst. This method permits the practical synthesis of valuable chiral N-allylated indoles, and avoids the N- or C-selectivity issue.
Similar content being viewed by others
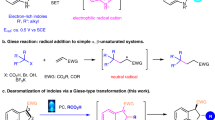

Introduction
The asymmetric synthesis of indoles is of great interest because of their prevalence in bioactive molecules1,2,3,4,5,6. In particular, indoles bearing a α-chiral carbon centre on the N are important structural motifs in natural products and pharmaceutical drugs (Fig. 1a)7,8,9,10,11,12. For this reason, extensive efforts have been undertaken to explore the catalytic asymmetric allylation of indoles13,14,15,16,17,18,19,20,21,22,23,24. However, selective N-allylation of indoles is a true challenge due to the high nucleophilicity of C3 of the indole nucleus and the weak acidity of the N–H bond (Fig. 1b)25,26. As a consequence, efficient strategies for the synthesis of N α-chiral allylic indoles are still rare. Recent advances were achieved upon installation of an electron-withdrawing substituent at C2 or C3 positions, which tempers the nucleophilicity at C3 and increases the acidity of the N–H bond22. In addition, a two-step protocol by allylation/oxidation of indolines could avoid C3 selectivity issue (Fig. 1c)24.

(a) Selected examples of biologically active N α-chiral indoles. (b) Selectivity issue for allylation of indoles. (c) Strategies for transition metal-catalysed asymmetric synthesis of N-allylic indoles. LG, leaving group; EWG, electron-withdrawing group; FI, Fischer indolization.
Potentially, chiral N1-allylic aryl hydrazine could give access to various chiral N-allylic indoles by employing a well-established Fischer indole synthesis27,28,29,30. This method would allow flexible construction of complex chiral N-allylic indoles starting from commercially accessible materials (ketones and aldehydes). Challenge towards the synthesis of chiral N1-allylic aryl hydrazines arises from the selectivity control: (1) N1 and N2 selectivity of aryl hydrazines31,32,33; (2) branched and linear selectivity of the allylic moiety; (3) enantioselectivity of the branched regioisomer. To address these issues, we envisioned that a transition metal-catalysed asymmetric N1-selective coupling of aryl hydrazines with terminal allenes34,35,36,37,38,39,40 could lead to the synthesis of chiral N1-allylic aryl hydrazines. The N1 and N2 selectivities at the aryl hydrazine may differentiate in the oxidative addition step, in which the more acidic N–H bond at N1 proceeds faster than the less acidic N–H bond at N2. Furthermore, combination of a suitable transition metal catalyst and a chiral ligand may allow to control branched selectivity and enantioselectivity.
Herein we report a rhodium-catalysed regio- and enantioselective coupling of aryl hydrazines with terminal allenes, which lead to the asymmetric synthesis of N-allylic indoles by following a Fischer indolization (Fig. 1c).
Results
Reaction optimization of aryl hydrazine allylation
To evaluate our assumption, our studies began with the coupling reaction of phenyl hydrazine and cyclohexylallene in the presence of [Rh(COD)Cl]2 (1.25 mmol%) and DPEphos (5.0 mmol%) in 1,2-dichloroethane at 80 °C. Surprisingly, the desired N1-selective branched product was isolated with a promising 77% yield as a single regioisomer (Table 1, entry 1). Encouraged by the high N1 and branched regioselectivities, we then tested a range of chiral bidentate phosphine ligands (Table 1, entries 2–10). The ligands Josiphos L and (R,R)-Diop led to low yield or poor enantioselectivity (Table 1, entries 2 and 3). After extensive screening (see Supplementary Table 1), we were pleased to observe that biaryl-type bisphosphine ligands led to high yield and promising enantiomeric excess (ee) (Table 1, entries 4–6). Increasing the steric effect of the Segphos-type ligand could significantly increase the enantioselectivity. The best ee was obtained with a bulky (S)-DTBM-Segphos ligand (Table 1, entries 6–8). Similarly, (R)-DTBM-Binap gave a comparable result (Table 1, entry 9). The enantiomeric purity could be enriched by a single recrystallization of the toluene sulfonic acid salt. Control experiments indicated that both rhodium catalyst and ligand are necessary for the coupling reaction of aryl hydrazine with allene to proceed (Table 1, entries 10 and 11).
Substrates scope of aryl hydrazine allylation
With the optimized conditions in hand, we then examined the scope of the addition of different aryl hydrazines with terminal allenes (Fig. 2). Various aryl hydrazines were coupled with cyclohexylallene in up to 93% isolated yield (1a) and up to 91% ee (1d–e). Practically, the N-allylated aryl hydrazines can be recrystallized from the corresponding tosylic acid salts to enrich the enantiomeric excess (1a–c). Several mono-substituted allenes were also tested (1h–l). Allenes bearing a phthaloyl-protected amine, an ester function and a silylether were suitable (1g–l).

[a]Isolated yield. [b]Determined by chiral HPLC. [c]ee after recrystallization from tosylic acid salt. [d]Reaction in 1.0 mmol scale. [e][Rh(cod)Cl]2 (2 mol%), L1 or L2 (8.0 mol%). [f]Reaction at 100 °C. [g]The NMR spectrum of 1j is not fully pure due to contamination of acetone (formation of hydrazone) during the purification. This problem can be avoided in the process of one-pot asymmetric synthesis of N-allylic indoles. Chp, cycloheptyl; HPLC, high-performance liquid chromatography; Phth, phaloyl; TBS, tert-butylsilyl.
One-pot asymmetric synthesis of N-allylic indoles
To investigate the compatibility of our strategy in the synthesis of N-allylic indoles via a one-pot process, the crude reaction mixture of the coupling step (1a) was subjected directly for the Fischer indolization with cyclohexanone in acetic acid at 70 °C. The desired N-allylic indole 2a was obtained in 87% isolated yield over two steps with retention of the enantiomeric purity. Variation with other aryl hydrazines and allenes using this one-pot process led to the synthesis of the corresponding N-allylic indoles in up to 90% yield and up to 91% ee (2b–f). Furthermore, aldehydes, phenyl-substituted ketones as well as a dihydro-2H-thiopyran-4(3H)-one were well tolerated under standard conditions (Fig. 3).

[a]Scope conditions of Fig. 2. [b]Reaction carried out at 100 °C.
To test the scalability and application for the synthesis of bioactive molecules, we applied the one-pot process for the late-stage indolization of (+)-testosterone acetate. To our delight, the desired indole product 3 was obtained in 59% yield and 17:1 diastereoselectivity in 1.06 gram scale, which indicates the practicality and usefulness of the method (Fig. 4).

[a][Rh(cod)Cl]2 (1.0 mol%), L2 (4.0 mol%), 1,2-dichloroethane (0.4 M), 80 °C, 19 h.
Mechanistic investigations
To probe the possible reaction mechanism, a control experiment of 1-methyl-1-phenylhydrazine with cyclohexylallene was conducted under optimized conditions (Fig. 5a). The reaction was sluggish and gave only traces of the N2-allylated product 4, which is in accord with the lower reactivity of N2 of the aryl hydrazine. Deuterium-labelling experiments with [D3]phenylhydrazine under optimized conditions displayed that deuterium was only incorporated in the internal position of the double bond (Fig. 5b). Stoichiometric reaction of phenylhydrazine with [{Rh(COD)Cl}2] and DPEphos in CDCl3 was monitored by NMR spectroscopy. After 5 min at room temperature, the 1H NMR spectrum (263 K) showed a major rhodium hydride species at δ=−15.4 p.p.m. (1JRh-H=14 Hz), which is indicative of the oxidative addition of the N–H bond to rhodium (Fig. 5c).

(a) Control experiment with 1-methyl-1-phenylhydrazine. (b) Isotopic-labelling experiment with [D3]phenylhydrazine. (c) Stoichiometric reaction of phenylhydrazine with catalysts. (d) Proposed mechanism.
On the basis of these observations, the following mechanism can be proposed (Fig. 5d). Oxidative addition of the phenyl hydrazine to Rh(I) generates Rh(III) complex (A or A′)41. The oxidative addition step favours the formation of intermediate A because of the higher acidity of N–H bond of N1 than N2. Hydrometalation of the less-substituted double bond could generate π-allyl-Rh (or δ-allyl-Rh) complex B (or B′)42,43,44,45, which could generate the desired branched N-allylic aryl hydrazine 1a via reductive elimination. The N-selectivity was determined within the oxidative addition step46.
Discussion
We have developed the enantioselective N-selective coupling of aryl hydrazines with allenes via a rhodium(I)/DTBM-Segphos or rhodium(I)/DTBM-Binap catalyst system, which allowed the asymmetric synthesis of various valuable N-allylic indoles by following a one-pot Fischer indolization. N-selective allylation of aryl hydrazines using alkynes, target-oriented synthesis, and mechanistic investigations are currently underway in our laboratory and will be reported in due course.
Methods
Allylation of aryl hydrazines
To a screw-cap Schlenk tube was added [Rh(cod)Cl]2 (0.005 mmol, 1 mol%), L1 or L2 (0.02 mmol, 4 mol%), aryl hydrazine (0.5 mmol, 1.0 equiv.), 1,2-dichloroethane (0.4 M) and allene (0.75 mmol, 1.5 equiv.). The Schlenk tube was sealed and the mixture was stirred for 19 h at 80 °C (or 100 °C). After cooling to room temperature, the solvent was removed by rotary evaporation. The crude product was purified by flash column chromatography to obtain the corresponding allylic hydrazine.
One-pot asymmetric synthesis of N-allylic indoles
To the reaction mixture of allylation of hydrazine was added ketone or aldehyde (0.55 mmol, 1.1 equiv.), and the mixture was stirred for half hour to form the corresponding hydrazine, then solvent was removed under reduced pressure. To the residue was added acetic acid (2.0 ml, 0.25 M), and the reaction mixture was stirred for 3–18 h at 70 °C (or 100 °C). The volatiles were removed by rotary evaporation and the crude reaction mixture was purified by flash column chromatography. The ee of each product was determined by HPLC analysis using chiral stationary phases. All new compounds were fully characterized. For NMR, high resolution mass spectrometry (HRMS) analysis and HPLC traces of the compounds in this article, see Supplementary Figs 1–53. General information, materials, synthesis and characterization of compounds in this article (1a–l, 2a–i, 3, 4 and 5), and experimental part for mechanistic investigations see Supplementary Methods.
Additional information
How to cite this article: Xu, K. et al. Asymmetric synthesis of N-allylic indoles via regio- and enantioselective allylation of aryl hydrazines. Nat. Commun. 6:7616 doi: 10.1038/ncomms8616 (2015).
References
Gul, W. & Hamann, M. T. Indole alkaloid marine natural products: an established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 78, 442–453 (2005).
Pasquali, G., Porto, D. D. & Fett-Neto, A. G. Metabolic engineering of cell cultures versus whole plant complexity in production of bioactive monoterpene indole alkaloids: recent progress related to old dilemma. J. Biosci. Bioeng. 101, 287–296 (2006).
Lewis, S. E. Recent advances in the chemistry of macroline, sarpagine and ajmaline-related indole alkaloids. Tetrahedron 62, 8655–8681 (2006).
O'Connor, S. E. & Maresh, J. J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat. Prod. Rep. 23, 532–547 (2006).
Higuichi, K. & Kawasaki, T. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat. Prod. Rep. 24, 843–868 (2007).
Kochanowska-Karamyan, A. J. & Hamann, M. T. Marine indole alkaloids: potential new drug leads for the control of depression and anxiety. Chem. Rev. 110, 4489–4497 (2010).
Maincent, P., Verge, R. L., Sado, P., Couvreur, P. & Devissaguet, J. P. Disposition kinetics and oral bioavailability of vincamine-loaded polyalkyl cyanoacrylate nanoparticles. J. Pharm. Sci. 75, 955–958 (1986).
Herzberg, U., Eliav, E., Bennett, G. J. & Kopin, I. J. The analgesic effects of R(+)-WIN 55,212-2 mesylate, a high affinity cannabinoid agonist, in a rat model of neuropathic pain. Neurosci. Lett. 221, 157–160 (1997).
Rahbæk, L. & Christophersen, C. Marine alkaloids. 19. Three new alkaloids, securamines E-G, from the marine bryozoan Securiflustra securifrons. J. Nat. Prod. 60, 175–177 (1997).
Vepsäläinen, J. J., Auriola, S., Tukiainen, M., Ropponen, N. & Callaway, J. C. Isolation and characterization of yuremamine, a new phytoindole. Planta Med. 71, 1053–1057 (2005).
Fernandez, L. S., Buchanan, M. S., Carroll, A. R., Feng, Y. J. & Quinn, R. J. Flinderoles A-C: antimalarial bis-indole alkaloids from Flindersia species. Org. Lett. 11, 329–332 (2009).
Vallakati, R., Smuts, J. P., Armstrong, D. W. & May, J. A. On the biosynthesis and optical activity of the flinderoles. Tetrahedron Lett. 54, 5892–5894 (2013).
Kimura, M., Futamata, M., Mukai, R. & Tamaru, Y. Pd-catalyzed C3-selective allylation of indoles with allyl alcohols promoted by triethylborane. J. Am. Chem. Soc. 127, 4592–4593 (2005).
Zaitsev, A. B. et al. Fast and highly regioselective allylation of indole and pyrrole compounds by allyl alcohols using Ru-sulfonate catalysts. J. Am. Chem. Soc. 130, 11604–11605 (2008).
Liu, W., He, H., Dai, L. & You, S. Ir-catalyzed regio- and enantioselective friedel-crafts-type allylic alkylation of indoles. Org. Lett. 10, 1815–1818 (2008).
Sundararaju, B. et al. Ruthenium (IV) complexes featuring P,O-chelationg ligands: regioselective substitution directly from allylic alcohols. Angew. Chem. Int. Ed. 49, 2782–2785 (2010).
Jiao, L., Herdtweck, E. & Bach, T. Pd(II)-catalyzed regioselective 2- alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications. J. Am. Chem. Soc. 134, 14563–14572 (2012).
Sevov, C. S. & Hartwig, J. F. Iridium-catalyzed intermolecular asymmetric hydroheteroarylation of bicycloalkenes. J. Am. Chem. Soc. 135, 2116–2119 (2013).
Trost, B. M., Krische, M. J., Berl, V. & Grenzer, E. M. Chemo-, regio-, and enantioselective Pd-catalyzed allylic alkylation of indolocarbazole pro-aglycons. Org. Lett. 4, 2005–2008 (2002).
Cui, H. et al. Chemoselective asymmetric N-allylic alkylation of indoles with Morita-Baylis-Hillman carbonates. Angew. Chem. Int. Ed. 48, 5737–5740 (2009).
Luzung, M. R., Lewis, C. A. & Baran, P. S. Direct, chemoselective N-tert-prenylation of indoles by C-H functionalization. Angew. Chem. Int. Ed. 48, 7025–7029 (2009).
Stanley, L. M. & Hartwig, J. F. Iridium-catalyzed regio- and enantioselective N-allylation of indoles. Angew. Chem. Int. Ed. 48, 7841–7844 (2009).
Trost, B. M., Osipov, M. & Dong, G. Palladium-catalyzed dynamic kinetic asymmetric transformation of vinyl aziridines with nitrogen heterocycles: rapid access to biologically active pyrroles and indoles. J. Am. Chem. Soc. 132, 15800–15807 (2010).
Liu, W., Zhang, X., Dai, L. & You, S. Asymmetric N-allylation of indoles through the iridium-catalyzed allylic alkylation/oxidation of indolines. Angew. Chem. Int. Ed. 51, 5183–5187 (2012).
Lakhdar, S. et al. Nucleophilic reactivities of indoles. J. Org. Chem. 71, 9088–9095 (2006).
Otero, N., Mandado, M. & Mosquera, R. A. Nucleophilicity of indole derivatives: activating and deactivating effects based on proton affinities and electron density properties. J. Phys. Chem. A 111, 5557–5562 (2007).
Wagaw, S., Yang, B. H. & Buchwald, S. L. A palladium-catalyzed strategy for the preparation of indoles: a novel entry into the fisher indole synthesis. J. Am. Chem. Soc. 1998, 120 6621–6622 (2006).
Boal, B. W., Schammel, A. W. & Garg, N. K. An interrupted fischer indolization approach toward fused indoline-containing natural products. Org. Lett. 11, 3458–3461 (2009).
Müller, S., Webber, M. J. & List, B. The catalytic asymmetric fischer indolization. J. Am. Chem. Soc. 133, 18534–18537 (2011).
Gore, S., Baskaran, S. & König, B. Fischer indole synthesis in low melting mixtures. Org. Lett. 14, 4568–4571 (2012).
Ragnarsson, U. et al. Acidity of di- and triprotected hydrazine derivatives in dimethyl sulfoxide and aspects of their alkylation. J. Org. Chem. 70, 5916–5921 (2005).
Bredihhin, A., Groth, U. M. & Mäeorg, U. Efficient methodology for selective alkylation of hydrazine derivatives. Org. Lett. 9, 1097–1099 (2007).
Johns, A. M., Liu, Z. & Hartwig, J. F. Primary tert- and sec- allylamines via palladium-catalyzed hydroamination and allylic substitution with hydrazine and hydroxylamine derivatives. Angew. Chem. Int. Ed. 46, 7259–7261 (2007).
Zimmer, R., Dinesh, C., Nandanan, E. & Khan, F. A. Palladium-catalyzed reactions of allenes. Chem. Rev. 100, 3067–3125 (2000).
Trost, B. M., Jäkel, C. & Plietker, B. Palladium-catalyzed asymmetric addition of pronucleophiles to allenes. J. Am. Chem. Soc. 125, 4438–4439 (2003).
Nishina, N. & Yamamoto, Y. Gold-catalyzed intermolecular hydroamination of allenes with arylamines and resulting high chirality transfer. Angew. Chem. Int. Ed. 45, 3314–3317 (2006).
Kim, I. S. & Krische, M. J. Iridium-catalyzed hydrocarboxylation of 1,1-dimethylallene: byproduct-free reverse prenylation of carboxylic acids. Org. Lett. 10, 513–515 (2006).
Moran, J., Preetz, A., Mesch, R. A. & Krische, M. J. Iridium-catalyzed direct C-C coupling of methanol and allenes. Nat. Chem. 3, 287–290 (2011).
Koschker, P., Lumbroso, A. & Breit, B. Enantioselective synthesis of branched allylic esters via rhodium-catalyzed coupling of allenes with carboxylic acids. J. Am. Chem. Soc. 133, 20746–20749 (2011).
Xu, K., Thieme, N. & Breit, B. Unlocking the N2 selectivity of benzotriazoles: regiodivergent and highly selective coupling of benzotriazoles with allene. Angew. Chem. Int. Ed. 53, 7268–7271 (2014).
Ardizzoia, G. A. et al. Oxidative addition of N-H bonds to a metal center: synthesis, characterization, and crystal structure of new rhodium (III) hydrido-pyrazolate complexes. Inorg. Chem. 41, 610–614 (2002).
Choi, J., Osakada, K. & Yamamoto, T. Single and multiple insertion of arylallene into the Rh-H bond to give (π-allyl)rhodium complexes. Organometallics 17, 3044–3050 (1998).
Tran, N. & Cramer, N. Rhodium-catalyzed dynamic kinetic asymmetric transformations of racemic allens by the [3+2] annulation of aryl ketimines. Angew. Chem. Int. Ed. 52, 10630–10634 (2013).
Evans, P. A. & Nelson, J. D. Conservation of absolute configuration in the acyclic rhodium-catalyzed allylic alkylation reaction: evidence for an enyl (δ+π) organorhodium intermediate. J. Am. Chem. Soc. 120, 5581–5582 (1998).
Wucher, B., Moser, M., Schumacher, S. A., Rominger, F. & Kunz, D. First X-ray structure analyses of rhodium(III) η1–allyl complexes and a mechanism for allylic isomerization reactions. Angew. Chem. Int. Ed. 48, 4417–4421 (2009).
Gellrich, U. et al. Mechanistic investigations of the rhodium catalyzed propargylic CH activation. J. Am. Chem. Soc. 136, 1097–1104 (2014).
Acknowledgements
This work was supported by the DFG, the International Research Training Group ‘Catalysts and Catalytic Reactions for Organic Synthesis’ (IRTG 1038), the Fonds der Chemischen Industrie and the Krupp Foundation. We thank Umicore, BASF and Wacker for generous gifts of chemicals. K.X. thanks Dr Y.Z. Xia and Dr C.K. Li for helpful discussions.
Author information
Authors and Affiliations
Contributions
K.X. initiated the project, planned and carried out the initial optimization; K.X. and T.G. completed the experimental work and final characterizations; B.B. directed and coordinated the project; K.X wrote the manuscript with the assistance of the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-53, Supplementary Table 1, Supplementary Methods and Supplementary References (PDF 4325 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, K., Gilles, T. & Breit, B. Asymmetric synthesis of N-allylic indoles via regio- and enantioselective allylation of aryl hydrazines. Nat Commun 6, 7616 (2015). https://doi.org/10.1038/ncomms8616
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8616
This article is cited by
-
Enantioselective synthesis of N-alkylindoles enabled by nickel-catalyzed C-C coupling
Nature Communications (2022)
-
Graphene/MoS2/FeCoNi(OH)x and Graphene/MoS2/FeCoNiPx multilayer-stacked vertical nanosheets on carbon fibers for highly efficient overall water splitting
Nature Communications (2021)
-
Direct asymmetric N-propargylation of indoles and carbazoles catalyzed by lithium SPINOL phosphate
Nature Communications (2020)
-
Chromoselective access to Z- or E- allylated amines and heterocycles by a photocatalytic allylation reaction
Nature Communications (2019)
-
Green synthesis of NiFe LDH/Ni foam at room temperature for highly efficient electrocatalytic oxygen evolution reaction
Science China Materials (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
