
Abstract
The benzene unit, in its substituted forms, is a most common scaffold in natural products, bioactive molecules and polymer materials. Nearly 80% of the 200 best selling small molecule drugs contain at least one benzene moiety. Not surprisingly, the synthesis of substituted benzenes receives constant attentions. At present, the dominant methods use pre-existing benzene framework to install substituents by using conventional functional group manipulations or transition metal-catalyzed carbon-hydrogen bond activations. These otherwise impressive approaches require multiple synthetic steps and are ineffective from both economic and environmental perspectives. Here we report an efficient method for the synthesis of substituted benzene molecules. Instead of relying on pre-existing aromatic rings, here we construct the benzene core through a carbene-catalyzed formal [3+3] reaction. Given the simplicity and high efficiency, we expect this strategy to be of wide use especially for large scale preparation of biomedicals and functional materials.
Similar content being viewed by others
Introduction
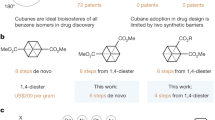
Multi-substituted benzenes are widely present in natural products. In industry, these benzene frameworks are nearly unavoidable in preparing most of today’s biomedicals, fine chemicals and polymer materials. The functions of the benzene-containing molecules are determined by the identity and substitution patterns of the substituents installed on the benzene unit. Exemplified in Fig. 1a are one natural product (salvadorin)1 and two synthetic bioactive molecules2,3 containing a benzene core bearing four substituents. Most synthetic methods in synthesizing such multi-substituted aromatics start with pre-existing benzene unit by replacing hydrogen with other functional groups. The classic approach relies on stepwise electrophilic substitution (such as Friedel–Crafts reaction) or electrophilic halogenation and successive transition metal-catalyzed couplings. However, regio-selectivity and chemo-selectivity normally require rather tedious functional group (including protecting group) manipulations. For example, the classic synthesis of a 2,4,6-trisubstituted benzoate needs the introduction of a temporary amine group to ensure selectivities in a key bromination reaction step4, and the overall synthesis requires over eight steps (Fig. 1b). Another approach for access to substituted benzenes is based on transition metal-catalyzed direct C–H activations5,6,7,8. While providing impressive shortcuts for benzene substitutions, this C–H activation method has its own limitations. For example, the presence of directing groups (for coordination with the metal catalyst) is often necessary and the instruction of multiple substituents is difficult (in part due to steric congestion). In a different direction for substituted benzene synthesis, the benzene core is newly formed. Representative methods include transition metal-catalyzed [2+2+2] or [4+2] reactions such as acetylene trimerizations developed by Reppe et al.9,10,11 In this cycloaddition approach, partial or complete intramolecular reaction is usually indispensable to ensure the regio-selectivity.

(a) examples of natural products and bioactive synthetic compounds containing multi-substituted benzene. (b) it took about eight steps for the classical substitution methods to synthesize a 2,4,6-trisubstituted benzoate. (c) transition metal-catalyzed C–H activation methods provides a shortcut (about five steps) for the synthesis. (d) this work: single key step reaction to afford tetra-substituted benzenes via organocatalyzed formal [3+3] cycloaddtion.
Here we report a new strategy for highly effective access to multi-substituted benzenes through the construction of the benzene core via a formal [3+3] cycloaddition reaction (Fig. 1d). Our approach uses enals readily prepared in three steps and unsaturated ketones as the starting material and N-heterocyclic carbene (NHC) as the organic catalyst12,13,14,15,16,17,18,19,20,21,22,23,24. It is a single-step reaction that affords tetra-substituted benzenes (2,4,6-trisubstituted benzoate and its analogues) with high yield. In comparison, previous approaches to this class of molecules typically need seven steps with less than 10% overall yields25 (Fig. 1d). A plausible pathway of our NHC-catalyzed [3+3] cycloaddition reaction involving formal α-, and γ-carbon activations of enal is illustrated in Fig. 2. Briefly, addition of the carbene catalyst to the aldehyde moiety of enal followed by deprotonation forms Breslow intermediate I26,27,28. This process is followed by oxidative transformation29,30,31,32,33, and former enal γ-CH deprotonation33 leads to vinyl enolate intermediate III. Notably, similar vinyl enolate intermediate could also be accessed from ketenes34 by Ye or esters in our laboratory35. Nucleophilic Michael-type addition of the γ-carbon of III to enone 2 affords intermediate IV bearing a NHC-bound α,β-unsaturated ester moiety. Subsequent γ-CH deprotonation of IV lead to dienolate intermediate V that undergoes an intramolecular aldol to form a cyclic intermediate VI. Intramolecular ester formation with the release of NHC catalyst yields a bicyclic adduct VII containing a four-membered β-lactone. Spontaneous decarboxylation36,37,38,39 and oxidation40,41,42 of VII effectively affords benzene product bearing four substituents in predictable substitution patterns. Decarboxylation of β-lactone fused with a six-membered ring similar to intermediate VII is a highly effective process, as exemplified in Lupton’s [4+2] reaction via carbene catalysis39.

Addition of carbene catalyst to aldehyde followed by oxidative transformation and γ-CH deprotonation leads to intermediate III, which undergoes Michael addition to 2 and gives intermediate IV. Subsequent γ-CH deprotonation of IV lead to V, after intramolecular adol reaction, decarboxylation and oxidation, finally gives a benzene product bearing four substituents in predictable substitution patterns. Mes, 2,4,6-trimethylphenyl. Cat., catalyst.
Results
Reaction optimization
We started by using enal 1a and enone 2a as the model substrates, in the presence of 2 equiv. quinone 4 as an oxidant29,30,31,32,33 and Cs2CO3 as a base. No formation of the proposed benzene 3a was observed in the absence of an NHC precatalyst (Table 1, entry 1). The N-methyl imidazolium NHC A (ref. 43) and N-phenyl imidazolium B could not initiate the reaction (Table 1, entries 2–3). We then found that with N-Mes imidazolium C (ref. 44) as the NHC precatalyst, the proposed product 3 was formed in 88% isolated yield (Table 1, entry 4). Triazolium-based NHCs behaved similarly as the imidazolium catalysts: triazolium D (ref. 45) (with a N-phenyl substituent) could not catalyze the reaction; while the use of triazolium E (ref. 46) with a N-mesityl substituent could lead the formation of 3 in 47% yield (Table 1, entries 5–6). Thiazolium-based NHCs F (ref. 47) or G (ref. 48) could not initiate the reaction (Table 1, entries 7–8). We then evaluated the effects of solvents and bases. Although the combination of tetrahydrofuran and Cs2CO3 was optimal, other common organic solvents (such as CH2Cl2, toluene, CH3CN and DMF) and organic/inorganic bases (such as Et3N, DBU, tBuOK and K2CO3) could also be used (see the Supplementary Table 1). Further investigation showed that the catalyst loading of C could be decreased to 5 mol% with acceptable 76% yield (Table 1, entry 9).
Scope of enal substrates
With an acceptable reaction condition in hand (Table 1, entry 9), the scope of the reaction was evaluated. To demonstrate broader synthetic utility of this method, we chose enone 2b bearing an alkene group (amenable for further transformation) as a model enone substrate to study the generality of the enal substrates (Fig. 3, products 3b–j). Both electron-donating (products 3c–d, 3f) and electron-withdrawing group (products 3e, 3g) at the p- position (products 3c–e) or m- position (products 3f–g) of the β-phenyl group were well-tolerated. Replacement of the β-phenyl substituent with a naphthyl (product 3h) or heteroaryl unit (products 3i–j) had little effect on the reaction outcome. It is worth to note that E- or Z-isomer of enal 1 gave essentially the same yields, so a mixture of E-/Z-enals can be directly used.

(a) Examples of different enals: study of enal substrate generality with ketone 2b as a model enone substrate. (b) Examples of different enones: study of enones scope with aldehydes 1a (Ar=C6H5) and 1b (Ar=4-OCH3-C6H4) as model nucleophile. Ph, phenyl; Me, methyl; Et, ethyl.
Scope of enone substrates
With aldehydes 1a and 1b (Ar=4-OCH3-C6H4) as model nucleophile, the scope of enones was also examined. As shown in Fig. 3 (products 3k–z1), in nearly all cases, the reaction proceeded smoothly at room temperature to give polysubstituted benzenes in moderate to good yields. Notably, the E-/Z-configuration of enone 2 did not affect the reaction outcomes either (for example, see product 3k), which greatly simplify substrate preparation process. Both electron-rich (products 3p–q) or electron-deficient (products 3o and 3r) aromatic groups, as well as vinyl groups (products 3k–m) were all tolerated in the β-substituent (R2) of the enone substrates. α-Substituent (R3) of the enone substrates can be electron-deficient units such as acyl(3m, 3r, 3v–3w), ester (3k–3l, 3n–q) or nitro (3y) groups. Substituent in the carbonyl group of enones 2 (R4) can be a different alkyl group including methyl (3k–q, 3r–s), ethyl (3t and 3w), isopropyl (3u) or trifluoromethyl (3z–z1) group.
It is important to note that these polysubsituted benzene molecules were difficult to prepare previously. For example, previous method for the synthesis of benzoate 3o required seven steps with less than 10% overall yields25; previous synthesis of 3p and 3q needed a three-step reaction with 35 and 39% overall yield49, respectively. Our method also provides effective access to aromatic molecules with trifluoromethyl (CF3) substituent. In organic synthesis, regioselective C–H trifluoromethylation50,51 or methylation52,53,54 of aryl molecules still remains challenging despite the rapid development in recent years.
Product transformation
We next demonstrated effective transformation of our catalytic reaction products to bicyclic and multicylic aromatic molecules that are found as a key scaffold in natural products and functional synthetic molecules. For example, the multisubsituted benzene adduct 3b could be transformed to indene 7 (ref. 55) via reduction followed by Lewis acid-mediated cyclization; 2,4,6-trisubstituted benzoate 3n can be transformed to fluorenone 5 (refs 56, 49) or isoindolone 6 (ref. 57) via straightforward processes (Fig. 4).

(a) 3n can be transfomed to fluorenone 5 or isoindolone 6 via straightforward processes. (b) 3b could be transformed to indene 7 via reduction followed by Lewis acid-mediated cyclization. NBS, N-bromosuccinimide.
Discussion
In summary, we have developed a NHC organocatalytic strategy for the formal [3+3] construction of multi-substituted benzenes. Our method directly employs commercially available or easily accessible enals and enones to construct benzene framework with excellent regioselectivities, offering useful insights into the design of concise synthetic strategies for complex molecules. Further studies regarding reaction mechanisms, synthetic applications and axial chirality controls (for the formation of substituted benzene products) are under progress in our laboratory.
Methods
Materials
For 1H, 13C and 19F NMR spectra of compounds in this manuscript, see Supplementary Figs 1–32. For details of the synthetic procedures, see Supplementary Methods.
Synthesis of 3
Under N2 atmosphere, a solution of enal (0.1 mmol), enone (0.1 mmol), oxidant 4 (82 mg, 0.2 mmol), Cs2CO3 (48.7 mmg, 0.15 mmol) and imidazolium C (1.7 mg, 0.005 mmol) in 1.0 ml tetrahydrofuran was stirred at room temperature for 8 h. The mixture was concentrated under reduced pressure and purified by silica gel column chromatography to afford the corresponding product 3.
Additional information
How to cite this article: Zhu, T. et al. Benzene construction via organocatalytic formal [3+3] cycloaddition reaction. Nat. Commun. 5:5027 doi: 10.1038/ncomms6027 (2014).
References
Mahmood, T., Ahmed, E. & Malik, A. Structure determination of salvadorin, a novel dimeric dihydroisocoumarin from salvadora oleoides, by NMR spectroscopy. Magn. Reson. Chem. 43, 670–672 (2005).
Stokker, G. E., Alberts, A. W., Gilfillan, J. L., Huff, J. W. & Smith, R. L. 3-hydroxy-3- methylglutaryl-coenzyme A reductase inhibitors. 5. 6-(Fluoren-9-yl)-and 6(Fluoren-9-ylidenyl)-3, 5-dihydroxyhexanoic acids and their lactone derivatives. J. Med. Chem. 29, 852–855 (1986).
Ortar, G. et al. 3-ylidenephthalides as a new class of transient receptor potential channel TRPA1 and TRPM8 modulators. Bioorg. Med. Chem. Lett. 23, 5614–5618 (2013).
Robison, M. M. & Robison, B. L. 2,4,6-tribromobenzoic acid. Org. Synth. 36, 94–97 (1956).
Cossy, J. & Arseniyadis, S. Modern Tools for the Synthesis of Complex Bioactive Ch. 1,1–32Wiley (2012).
Wencel-Delord, J., Dröge, T., Liu, F. & Glorius, F. Towards mild metal-catalyzed C–H bond activation. Chem. Soc. Rev. 40, 4740–4761 (2011).
Arockiam, P. B., Bruneau, C. & Dixneuf, P. H. Ruthenium (II)-catalyzed C-H bond activation and functionalization. Chem. Rev. 112, 5879–5918 (2012).
Engle, K. M., Mei, T.-S., Wasa, M. & Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C-H functionalization reactions. Acc. Chem. Res. 45, 788–802 (2012).
Reppe, W., Schlichting, O., Klager, K. & Toepel, T. Cyclisierende polymerisation von acetylen I über cyclooctatetraen. Justus Liebigs Ann. Chem. 560, 1–92 (1948).
Domínguez, G. & Pérez-Castells, J. Recent advances in [2+2+2] cycloaddtion reactions. Chem. Soc. Rev. 40, 3430–3444 (2011).
Hoye, T. R., Baire, B., Niu, D., Willoughby, P. H. & Woods, B. P. The hexadehydro-Diels-Alder reaction. Nature 490, 208–212 (2012).
Enders, D. & Balensiefer, T. Nucleophilic carbenes in asymmetric organocatalysis. Acc. Chem. Res. 37, 534–541 (2004).
Zeitler, K. Extending mechanistic routes in heterazolium catalysis-promising concepts for versatile synthetic methods. Angew. Chem. Int. Ed. 44, 7506–7510 (2005).
Marion, N., Diez-Gonzalez, S. & Nolan, S. P. N-heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 46, 2988–3000 (2007).
Nair, V., Vellalath, S. & Babu, B. P. Recent advances in carbon-carbon bond-forming reactions involving homoenolates generated by NHC catalysis. Chem. Soc. Rev. 37, 2691–2698 (2008).
Vora, H. U. & Rovis, T. Asymmetric N-heterocyclic Carbene (NHC) catalyzed acyl anion reactions. Aldrichim. Acta 44, 3–11 (2011).
Douglas, J., Churchill, G. & Smith, A. D. NHCs in asymmetric organocatalysis: recent advances in azolium enolate generation and reactivity. Synthesis. (Mass). 44, 2295–2309 (2012).
Phillips, E. M., Chan, A. & Scheidt, K. A. Discovering new reactions with N-heterocyclic carbene catalysis. Aldrichim. Acta 42, 55–66 (2009).
Izquierdo, J., Hutson, G. E., Cohen, D. T. & Scheidt, K. A. A continuum of progress: applications of N-hetereocyclic carbene catalysis in total synthesis. Angew. Chem. Int. Ed. 51, 11686–11698 (2012).
Ryan, S. J., Candish, L. & Lupton, D. W. Acyl anion free N-heterocyclic carbene organocatalysis. Chem. Soc. Rev. 42, 4906–4917 (2013).
Chiang, P.-C. & Bode, J. W. N-mesityl substituted chiral triazolium salts: opening a new world of N-heterocyclic carbene catalysis. TCI Mail 149, 2–17 (2011).
Bode, J. W. Carbene catalysis: an internal affair. Nat. Chem. 5, 813–815 (2013).
Bugaut, X. & Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 41, 3511–3522 (2012).
Hopkinson, M. N., Richter, C., Schedler, M. & Glorius, F. An overview of N-heterocyclic carbenes. Nature 510, 485–496 (2014).
Robl, J. A. A new and versatile route for the synthesis of highly substituted benzenoids. Tetrahedron Lett. 31, 3421–3424 (1990).
Teles, J. H. et al. Benzoin-type condensations of formaldehyde catalyzed by stable carbenes. Helv. Chim. Acta 79, 61–83 (1996).
Miyashita, A. et al. Synthesis and reactivities of 1,3-dimentyl-2-(α-hydroxybenzyl)imidazolium and 1,3-dimentyl-2-(α- hydroxybenzyl)benzimidazolium iodides. Heterocyles 44, 417–426 (1997).
Collett, C. J. et al. Mechanistic insights into the trazolylidene-catalysed Stetter and benzoin reactions: role of the N-aryl substituent. Chem. Sci. 4, 1514–1522 (2013).
Sarkar, S. D. & Studer, A. NHC-catalyzed michael addition to α,β-unsaturated aldehydes by redox activation. Angew. Chem. Int. Ed. 49, 9266–9269 (2010).
Rong, Z.-Q., Jia, M.-Q. & You, S.-L. Enantioselective N-heterocyclic carbene-catalyzed michael addition to α,β-unsaturated aldehydes by redox oxidation. Org. Lett. 13, 4080–4083 (2011).
Zhao, X., Ruhl, K. E. & Rovis, T. N-heterocyclic-carbene-catalyzed asymmetric oxidative hetero-Diels- Alder reactions with simple aliphatic aldehydes. Angew. Chem. Int. Ed. 51, 12330–12333 (2012).
Kravina, A. G., Mahatthananchai, J. & Bode, J. W. Enantioselective, NHC-catalyzed annulations of trisubstituted enals and cyclic N-sulfonylimines via α,β-unsaturated acyl azoliums. Angew. Chem. Int. Ed. 51, 9433–9436 (2012).
Mo, J., Chen, X. & Chi, Y. R. Oxidative γ-addition of enals to trifuoromethyl ketones: enantioselectivity control via Lewis acid/N-heterocyclic carbene cooperative catalysis. J. Am. Chem. Soc. 134, 8810–8813 (2012).
Shen, L.-T., Shao, P.-L. & Ye, S. N-heterocyclic carbene-catalyzed cyclization of unsaturated acyl chlorides and ketones. Adv. Synth. Catal. 353, 1943–1948 (2011).
Xu, J., Jin, Z. & Chi, Y. R. Organocatalytic enantioselective γ-aminoalkylation of unsaturated ester: access to pipecolic acid derivatives. Org. Lett. 15, 5028–5031 (2013).
Nair, V., Vellalath, S., Poonoth, M. & Suresh, E. N-heterocyclic carbene-catalyzed reaction of chalcones and enals via homoenolate: an efficient synthesis of 1,4,5-Trisubstituted cyclopentenes. J. Am. Chem. Soc. 128, 8736–8737 (2006).
Chiang, P.-C., Rommel, M. & Bode, J. W. α′-hydroxyenones as mechanistic probes and scope-expanding surrogates for α,β-Unsaturated aldehydes in N-heterocyclic carbene- catalyzed reactions. J. Am. Chem. Soc. 131, 8714–8718 (2009).
Cardinal-David, B., Raup, D. E. A. & Scheidt, K. A. Cooperative N-heterocyclic carbene/Lewis acid catalysis for highly stereoselective annulation reactions with homoenolates. J. Am. Chem. Soc. 132, 5345–5347 (2010).
Ryan, S. J., Candish, L. & Lupton, D. W. N-heterocyclic carbene-catalyzed (4+2) cycloaddtion/ decarboxylation of silyl dienol ethers with α,β-unsaturated acid fluorides. J. Am. Chem. Soc. 133, 4694–4697 (2011).
Izawa, Y., Pun, D. & Stahl, S. S. Palladium-catalyzed aerobic dehydrogeneation of substituted cyclohexanones to phenols. Science 333, 209–213 (2011).
Girard, S. A. et al. Pd-catalyzed synthesis of aryl amines via oxidative aromatization of cyclic ketones and amines with molecular oxygen. Org. Lett. 14, 5606–5609 (2012).
van Otterlo, W. A. & de Koning, C. B. Metathesis in the synthesis of aromatic compounds. Chem. Rev. 109, 3743–3782 (2009).
Ye, W., Cai, G., Zhuang, Z., Jia, X. & Zhai, H. One-step assembly of functionalized γ-butyrolactones from benzoins or benzaldehydes via an N-heterocyclic carbene-mediated tandem reaction. Org. Lett. 7, 3769–3771 (2005).
Burstein, C. & Glorius, F. Organocatalyzed conjugate umpolung of α,β-Unsaturated aldehydes for the synthesis of γ-butyrolactones. Angew. Chem. Int. Ed. 43, 6205–6208 (2004).
Reynolds, N. T., de Alaniz, J. R. & Rovis, T. Conversion of α-haloaldehydes into acylating agents by and internal redox reaction catalyzed by nucleophilic carbenes. J. Am. Chem. Soc. 126, 9518–9519 (2004).
Bode, J. W. & Sohn, S. S. N-heterocyclic carbene-catalyzed redox amidations of α-functionalized aldehydes with amines. J. Am. Chem. Soc. 129, 13798–13799 (2007).
Stetter, H. & Kulmann, H. Acyloin condensation by thiazolium ion catalysis: butyroin. Org. Synth. 62, 170–174 (1984).
Castells, J., Llitjos, H. & Moreno_Manas, M. Nitrobenzene aldehyde oxidations catalyzed by the conjugate bases of thiazolium ions. Tetrahedron Lett. 2, 205–206 (1977).
Reim, S., Lau, M. & Langer, P. Synthesis of fluorenones based on a ‘[3+3] cyclization/Suzuki cross-coupling/Friedel-Crafts acylation' strategy. Tetrahedron Lett. 47, 6903–6905 (2006).
Nagib, D. A. & Macmillan, D. W. C. Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis. Nature 480, 224–228 (2011).
Fujiwara, Y. et al. Practical and innate carbon-hydrogen functionalization of heterocycles. Nature 492, 96–99 (2012).
Schönherr, H. & Cernak, T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. Int. Ed. 52, 12256–12267 (2013).
Giri, R. et al. Palladium-Catalyzed Methylation and arylation of sp2 and sp3 C-H bonds in simple carboxylid acids. J. Am. Chem. Soc. 129, 3510–3511 (2007).
Rosen, B. R. et al. C-H functionalization logic enables synthesis of (+)-Hongoquercin A and related compounds. Angew. Chem. Int. Ed. 52, 7317–7320 (2013).
Antonio, D. S. et al. Preparation and pharmacological chracterization of trans-2-amino-5(6)-fluoro- 6(5)-hydroxy-1-phenyl-2,3-dihydro-1H-indenes as D2-like Dopamine receptor agonists. J. Med. Chem. 48, 2646–2654 (2005).
Perry, P. J. et al. 2,7-Disubstituted amdofluorenone derivatives as inhibitors of human telomerase. J. Med. Chem. 42, 2679–2684 (1999).
Lawson, E. C. et al. Nonpeptide Urotesin-II receptor antagonist: a new ligand class based on piperazino-phthalimide and piperazino-isoindolinone subunits. J. Med. Chem. 52, 7432–7445 (2009).
Acknowledgements
We acknowledge support by the Singapore National Research Foundation (NRF), the Singapore Economic Development Board (EDB), GlaxoSmithKline (GSK) and the Nanyang Technological University (NTU); China’s National Key programme for Basic Research (No. 2010CB 126105), the National Natural Science Foundation of China (No. 21132003) and the Guizhou University (China).
Author information
Authors and Affiliations
Contributions
T.Z. conducted most of the experiments; P.Z. and C.M. prepared substrates for the reaction scope evaluation; S.Y., B.-A.S. and Y.R.C. conceptualized and directed the project, and drafted the manuscript with the assistance from all co-authors. All authors contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-32, Supplementary Table 1, Supplementary Methods and Supplementary References (PDF 2223 kb)
Rights and permissions
About this article

Cite this article
Zhu, T., Zheng, P., Mou, C. et al. Benzene construction via organocatalytic formal [3+3] cycloaddition reaction. Nat Commun 5, 5027 (2014). https://doi.org/10.1038/ncomms6027
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms6027
This article is cited by
-
Electroredox carbene organocatalysis with iodide as promoter
Nature Communications (2022)
-
N-heterocyclic carbene-catalyzed arene formation reactions
Science China Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
