
Abstract
Ca2+ influx via voltage-dependent CaV1/CaV2 channels couples electrical signals to biological responses in excitable cells. CaV1/CaV2 channel blockers have broad biotechnological and therapeutic applications. Here we report a general method for developing novel genetically encoded calcium channel blockers inspired by Rem, a small G-protein that constitutively inhibits CaV1/CaV2 channels. We show that diverse cytosolic proteins (CaVβ, 14-3-3, calmodulin and CaMKII) that bind pore-forming α1-subunits can be converted into calcium channel blockers with tunable selectivity, kinetics and potency, simply by anchoring them to the plasma membrane. We term this method ‘channel inactivation induced by membrane-tethering of an associated protein’ (ChIMP). ChIMP is potentially extendable to small-molecule drug discovery, as engineering FK506-binding protein into intracellular sites within CaV1.2-α1C permits heterodimerization-initiated channel inhibition with rapamycin. The results reveal a universal method for developing novel calcium channel blockers that may be extended to develop probes for a broad cohort of unrelated ion channels.
Similar content being viewed by others


Introduction
High-voltage-activated Ca2+ (CaV1/CaV2) channels convert membrane electrical signals into Ca2+ influx that drives essential processes ranging from muscle contraction to synaptic transmission1. CaV1/CaV2 channels are hetero-multimers comprised minimally of any one of seven pore-forming α1 (CaV1.1–CaV1.4; CaV2.1–CaV2.3), four CaVβ (CaVβ1–β4) and four α2δ (α2δ1–α2δ4) subunits in a 1:1:1 ratio1,2. CaV1/CaV2 channel inhibition is an important or potential therapy for serious disorders including hypertension, neuropathic pain, stroke, Alzheimer’s and Parkinson’s disease3,4,5,6. L-type calcium (CaV1.1–1.4) channels are inhibited by dihydropyridines, phenylalkylamines and benzothiazepines7, whereas CaV2.1–2.3 channels are blocked by distinct venom toxins8. Nevertheless, the full potential of calcium channel blocker (CCB) therapy remains unrealized due to a lack of selective and tissue-specific small-molecule inhibitors for individual CaV1/CaV2 channel types. For example, clinically used L-type CCBs do not discriminate effectively among CaV1.1–CaV1.4 isoforms9. As L-type channels are widely expressed, this raises significant concerns for off-target effects when targeting specific CaV1 isoforms for neurological disorders such as Alzheimer’s and Parkinson’s diseases3,5. Genetically encoded intracellular-acting CCBs have the potential for a high therapeutic index because they can be expressed in a locally restricted manner2,10.
RGK (Rad/Rem/Rem2/Gem/Kir) GTPases are monomeric Ras-like G-proteins that powerfully inhibit all CaV1/CaV2 channels11,12,13. Two proof-of-concept experiments have demonstrated the potential powerful applications of RGK proteins as genetically encoded CCBs. First, local gene delivery of Gem to the atrioventricular node slowed atrioventricular nodal conduction and reduced heart rate in a porcine atrial fibrillation model10. Second, targeting Rem to caveolae in single cardiomyocytes permitted selective inhibition of CaV1.2 channels in this sub-cellular compartment14. The ability to inhibit CaV1/CaV2 channels in such a locally restricted manner at the whole organ or single-cell level cannot be achieved with traditional small-molecule CCBs. Ultimately, however, the potential applications of RGKs themselves as genetically encoded CCBs are limited because they do not discriminate among CaV1/CaV2 isoforms, and they have other diverse binding partners and biological functions including regulating the cytoskeleton11,15,16. These challenges may be overcome if it were possible to exploit the mechanism of action of RGKs to derive general principles for designing novel CCBs. Here we achieve this objective inspired by insights into how the RGK protein, Rem, inhibits CaV1/CaV2 channels.
Results
Differential tuning of CaV1/CaV2 channels by engineered Rem
Wild-type Rem targets to the plasma membrane using a polybasic C terminus tail and constitutively inhibits all CaV1 and CaV2 channel isoforms. Deleting the Rem C terminus tail (Rem265) ablates both membrane targeting and ICa inhibition. We previously discovered that fusing the C1 domain from protein kinase Cγ (C1PKC) to Rem1–265 enables it to be dynamically recruited to the plasma membrane with the phorbol ester, phorbol-12,13-dibutyrate (PdBu), resulting in concomitant inhibition of ICa. A surprising, but potentially fortuitous, feature of CaV channel inhibition by Rem265-C1PKC was that it displayed apparent selectivity, being effective for CaV1.2 and CaV2.2, but inert against CaV2.1 and CaV2.3. As a prelude to investigating whether the mechanism of Rem inhibition of CaV1/CaV2 channels could be exploited to develop a general method for developing genetically encoded CCBs, we explored the possibility of tuning the selectivity and potency of PdBu-inducible Rem-based CCBs by varying the relative positioning of the C1PKC motif in truncated Rem (Fig. 1a). When expressed in HEK 293 cells, all the truncated-Rem/C1PKC fusion proteins are basally cytosolic and are rapidly recruited to the plasma membrane with PdBu (Fig. 1b,c). Under basal conditions, robust whole-cell currents were recorded from cells co-expressing distinct CaV1/CaV2 channel subunits (α1+β) and the different Rem/C1PKC fusion constructs, enabling their selectivity and potency to be tested by adding 1 μM PdBu (Fig. 1d–g). Surprisingly, subtle differences in the relative position of C1PKC in truncated Rem dramatically impacted the comparative effectiveness of the resulting protein for CaV2.1 and CaV2.2 channels. For example, CFP-Rem1–265-C1PKCγ inhibited CaV2.2 (and CaV1.2) channels in response to PdBu, but was ineffective on CaV2.1 (Fig. 1d,h). In sharp contrast, simply shifting the C1PKC motif 15 residues closer to the G-domain produced CFP-Rem1–250-C1PKCγ, which permitted PdBu-induced inhibition of CaV2.1 (and CaV1.2) but was inert against CaV2.2 (Fig. 1e,h). Similarly, whereas C1PKC-YFP-Rem1–265 was effective for CaV2.2 but not CaV2.1 (Fig. 1f,h), C1PKC-Rem78–265-CFP had the opposite preference, with selectivity for CaV2.1 over CaV2.2 (Fig. 1g,h). By contrast with the tunable selectivity for CaV2.2/CaV2.1 channels, CaV1.2 was uniformly sensitive to all four truncated-Rem/C1PKC constructs. Overall, these results provide the novel observation that selectivity for distinct CaV2 channels can be engineered into Rem-based CCBs simply by altering the relative positioning of the inducible membrane-anchoring domain.

(a) Cartoons of wild-type (wt) Rem and engineered derivatives in which an inducible membrane-targeting C1PKC domain is placed in different positions. Wt Rem contains a guanine nucleotide-binding domain (G domain) appended by N- and C terminus extensions. The C terminus contains a polybasic sequence (MLS) that constitutively targets the protein to the plasma membrane. (b) Confocal images (top) and line scan plot (bottom) showing PdBu-induced translocation of CFP-Rem1–265-C1PKC from cytosol to the plasma membrane in a transfected HEK 293 cell. Scale bar, 4 μm. (c) Bar chart showing PdBu-induced increased plasma membrane to cytosol ratio for all four putative Rem-GEMIICCs. (d) Exemplar current waveforms for CaV2.2 (top), CaV2.1 (middle) and CaV1.2 (bottom) channels co-transfected with CFP-Rem1–265-C1PKC before (black trace) and shortly after (red trace) exposure to 1 μM PdBu. Scale bars, 400 pA, 10 ms. (e–g) Same format as (d), but with cells expressing CFP-Rem1–250-C1PKC, C1PKC-YFP-Rem1–265 and C1PKC-Rem78–265-CFP, respectively. (h) Population data showing differential impact of distinct Rem-GEMIICCs on CaV2.2, CaV2.1 and CaV1.2 channels. Data are means±s.e.m.
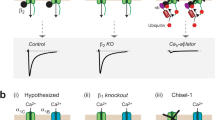
Conversion of auxiliary CaVβ3 into a CaV channel inhibitor
How does Rem binding to the plasma membrane result in CaV channel inhibition, and can this mechanism be exploited to identify a general method for developing genetically encoded CCBs? A critical clue for these questions is that in addition to membrane targeting (Fig. 1), binding to auxiliary CaVβ subunits is also required for CaV1.2 channel inhibition by CFP-Rem1–265-C1PKC17,18,19. CaVβs bind the intracellular domain I–II loop of pore-forming α1-subunits and promote channel trafficking to the plasma membrane, as well as modulating channel activation and inactivation gating20. To explain the dual requirement of membrane targeting and β binding for Rem inhibition of CaV1.2 open probability (Po), we hypothesized that though cytosolic variants of Rem constitutively associate with CaVβs in CaV1/CaV2 channel complexes, they are functionally silent, allowing the channel to gate and conduct current normally with depolarization (Fig. 2a, top left). By contrast, membrane-targeted Rem ‘tugs’ on CaVβ and by extension, the associated α1-subunit I–II loop, inducing a conformational change that closes the channel pore (Fig. 2a, top right). This allosteric model predicts it should be possible to convert the normally stimulatory CaVβ into a CaV channel inhibitor by directly inducing its association with the plasma membrane (Fig. 2a, bottom). We tested this idea by fusing C1PKC directly to the C terminus of CFP-β3 (Fig. 2b). CFP-β3-C1PKCγ is basally cytosolic but is rapidly recruited to the plasma (and nuclear) membrane with 1 μM PdBu (Fig. 2b, bottom left). When co-expressed with CaV2.2 (α1B), CFP-β3-C1PKCγ supported robust basal Ba2+ current (IBa), signifying a retained ability to promote α1-subunit membrane-trafficking and modulate gating (Fig. 2c). Remarkably, exposure to 1 μM PdBu resulted in a gradual decrease of IBa, peaking at 50% inhibition after 5 min (Fig. 2c, middle). The decrease in IBa extended across all relevant test pulse potentials, with no shift in the current density–voltage (I–V) relationship (Fig. 2c, right). Importantly, control cells expressing α1B+wild type (WT) β are not blocked by PdBu18 (Supplementary Fig. S1). Therefore, directly anchoring CFP-β3-C1PKCγ to the plasma membrane translates into IBa inhibition. This conversion of a normally stimulatory CaVβ subunit into a CCB provides strong evidence in support of the allosteric pore-closing model we propose for membrane-targeted Rem (Fig. 2a).

(a) Top, hypothesis for how the dual requirement for membrane-targeting and CaVβ-binding translates into Rem inhibition of CaV channels. Bottom, prediction for creating β-based CCBs. (b) Top, domain arrangement of CaVβ3 and sites of C1PKCγ placement in the C terminus of serially truncated CaVβ3 subunits. Bottom, PdBu-induced translocation of CFP-β3-C1PKCγ and CFP-β3[C16]-C1PKCγ from cytosol to plasma membrane. Scale bar, 5 μm. (c) Left, exemplar currents before (black trace) and after (red trace) exposure to PdBu in HEK 293 cell co-expressing CaV2.2α1B and CFP-β3-C1PKCγ. Scale bar, 0.5 nA, 10 ms. Middle, diary plot of IBa amplitude before (black squares) and after (red squares) exposure to 1 μM PdBu. Right, population I–V curves before (black triangles) and after (red triangles) 1 μM PdBu in cells expressing CFP-β3-C1PKCγ. Data are means±s.e.m, n=6 for each point. (d,e) Data for CFP-β3[C0]-C1PKCγ and CFP-β3[C16]-C1PKCγ, respectively; same format as (c), n=6 for each point in I–V plot. *P<0.05 compared with before PdBu data by two-tailed Student’s paired t-test. (f) Normalized ICa inhibition in HEK 293 cells expressing recombinant CaV2.2 (white triangles), CaV2.1 (black squares) or CaV1.2 (white squares) channels reconstituted with the distinct β-CCBs. Data are means±s.e.m, n=6 for each point. *significantly different from CFP-β3-C1PKCγ using one-way ANOVA and Bonferroni test. #P<0.05 compared with CaV2.2 data (blue line) by two-tailed Student’s paired t-test.
CaVβs have a conserved core comprised of a src homology 3 (SH3) and guanylate kinase (GK)-like domains separated by a variable HOOK domain, and flanked by variable-length unstructured N- and C termini21,22,23. An α1-binding pocket in CaVβ GK binds a conserved 18-residue α1 interaction domain in the α1-subunit I–II loop21,22,23,24. We hypothesized that placing C1PKCγ closer to GK would result in a more potent, and possibly, kinetically faster β3-based CCB. This is because the long and presumably floppy CaVβ C terminus might be expected to introduce some slackness in the putative PdBu-induced channel conformational change (Fig. 2a). To test this idea we generated a series of constructs in which the relative distance between C1PKCγ and GK was systematically varied by serial truncations of the long, unstructured β3 C terminus (Fig. 2b). The most extreme case featured C1PKCγ placed immediately downstream of GK, with no intervening β3 C terminus sequence, generating CFP-β3[C0]-C1PKCγ. Cells co-expressing α1B+CFP-β3[C0]-C1PKCγ displayed robust basal IBa, and exposure to 1 μM PdBu resulted in a strong (80%), rapid-onset inhibition of current (Fig. 2d). A construct in which C1PKCγ was separated from GK by 16 residues of the C terminus, CFP-β3[C16]-C1PKC, displayed the deepest PdBu-induced inhibition (90% inhibition at +10 mV test potential), with kinetics of onset intermediate between CFP-β3-C1PKCγ and CFP-β3[C0]-C1PKCγ (Fig. 2e). Population data from experiments using distinct variable-C terminus-length constructs revealed a robust 60–90% PdBu-induced inhibition of CaV2.2 for β3-CCBs with C termini ≤64 residues, some of which were significantly different from the 50% inhibition seen with full-length β3 (Fig. 2f, *P<0.05 compared with CFP-β3-C1PKCγ, by one-way ANOVA and Bonferroni test, n=6 for each point). Cells expressing α1B+CFP-β3[C16]-C1PKC+α2δ-1 were similarly inhibited by PdBu, indicating that the α2δ-1 subunit does not prevent this effect (Supplementary Fig. S1). To determine whether this mechanism of inhibition could be generalized to other CaV channels, we also assessed the efficacy of distinct β3-CCBs on CaV2.1 and CaV1.2 channels (Fig. 2f, Supplementary Figs S2 and S3). We found that the phenomenon was indeed evident in these other channels—for CaV1.2 the inhibition profile conferred by different β3-CCBs was similar to CaV2.2 (Fig. 2f, right), whereas CaV2.1 displayed a solid but significantly different inhibition pattern from CaV2.2 (Fig. 2f, middle, #P<0.05 compared with CaV2.2 inhibition, two-tailed unpaired t-test, n=6 for each point). Regarding the mechanism of inhibition of the β3-CCBs, we found that PdBu markedly decreased whole-cell current without affecting gating currents (Supplementary Fig. S4). This suggests a selective reduction in either channel Po or single-channel conductance, with no change in the number of channels at the cell surface, similar to what we previously found for PdBu-induced Rem-based CCBs18.
The effectiveness of membrane-targeted β3-CCBs in blocking CaV1/CaV2 channels was surprising given that two CaVβ isoforms, β2a and β2e, natively localize to the plasma membrane via their N termini, but nevertheless, yield robust IBa when reconstituted with α1-subunits25,26. Moreover, artificially introducing membrane-targeting domains to N termini of cytosolic CaVβs does not compromise their ability to reconstitute functional channels with α1-subunits27,28. One possibility for the discrepancy is that the polarity of the membrane-targeting domain on CaVβ is important for the impact on channel gating. We examined this idea by placing C1PKCγ on CaVβ3 N terminus and testing its effectiveness as a CCB. C1PKC-β3-CFP reconstituted robust IBa when co-expressed with CaV2.2α1B, but exposure to PdBu had minimal impact on IBa (Supplementary Fig. S5), suggesting polarity of the membrane-targeting domain is important for β-CCB efficacy.
General method for converting α1-binding proteins into CCBs
The β3-CCB results are consistent with a model in which membrane-targeted Rem uses CaVβ as a ‘handle’ to alter the conformation of α1-subunit I–II loop in a manner that closes the channel pore. As intracellular loops and termini of CaV1/CaV2 channels (Fig. 3a) engage in numerous protein–protein interactions, both common and unique, we speculated that other α1-binding proteins might be similarly used as ‘handles’ to manipulate channel gating. If so, this would reveal a generalized principle for designing novel genetically encoded CCBs. To test this premise, we focused on three different proteins (14-3-3, Ca2+-calmodulin-dependent protein kinase II and calmodulin) known to bind intracellular domains of individual CaV1/CaV2 channels29,30,31,32,33,34. Wild-type 14-3-3ε binds CaV2.2α1B C terminus and modulates channel inactivation properties31. To determine whether 14-3-3 could be converted into a small-molecule-regulated CaV2.2 inhibitor, we generated C1PKCγ-mCherry-14-3-3ε, which is normally cytosolic but rapidly translocates to the plasma membrane with PdBu (Fig. 3b). Cells co-expressing CaV2.2 (α1B+β) and C1PKC-mCherry-14-3-3 displayed robust basal IBa, and exposure to 1 μM PdBu caused a rapid 60% inhibition of ICa amplitude (Fig. 3c). Similar results were obtained with CaV2.1 and CaV1.2, respectively, indicating that these channels also interact with 14-3-3ε (Fig. 3c). To our knowledge, it was not previously known that CaV1.2 bound 14-3-3 proteins. Replacing C1PKCγ with an 18-residue palmitoylated membrane-targeting peptide (mem) from neuromodulin generated mem-mCherry-14-3-3ε, which constitutively targeted to the plasma membrane (Fig. 3d). Remarkably, mem-mCherry-14-3-3ε resulted in strong constitutive inhibition of CaV2.2, CaV2.1 and CaV1.2 channels across all test voltages (Fig. 3e). Control cells expressing mCherry-14-3-3 with CaV2.2 displayed neither inducible nor constitutive IBa inhibition (Supplementary Fig. S6).

(a) Schematic of CaV channel α1 subunit. Four homologous domains (I–IV) each with six transmembrane segments are joined by intracellular loops and bracketed by cytoplasmic N- and C termini. Various proteins bind CaVα1 intracellular domains including CaVβ, 14-3-3, CaM kinase II and calmodulin. (b) Confocal images showing PdBu-induced translocation of C1PKCγ-mCherry-14-3-3 from the cytosol to the plasma membrane. Scale bar, 4 μm. (c) Diary plots and population bar charts showing PdBu-induced inhibition of CaV2.2, CaV2.1, and CaV1.2 channels co-expressed with C1PKCγ-mCherry-14-3-3. Data are means±s.e.m. (d) Schematic and confocal image showing constitutive membrane targeting of mem-mCherry-14-3-3. Scale bar, 5 μm. (e) Population I–V curves showing constitutive inhibition by mem-mCherry-14-3-3 of CaV2.2 (black triangles, red triangles), CaV2.1 (black squares, red squares) and CaV1.2 (black circles, red circles) channels. Data are means±s.e.m, n=5 for each point. *P<0.05 compared with control data by two-tailed Student’s paired t-test. (f) PdBu-induced translocation of mCherry-CaMKII1–274,K42M-C1PKC from the cytosol to the plasma membrane. Scale bar, 7 μm. (g) Diary plots and population bar charts showing PdBu-induced inhibition of CaV2.2, CaV2.1 and CaV1.2 channels co-expressed with mCherry-CaMKII1–274,K42M-C1PKC. Data are means±s.e.m. (h) Schematic and confocal image showing constitutive membrane targeting of mCherry-CaMKII1–274,K42M-KRastail. Scale bar, 5 μm. (i) Constitutive inhibition of CaV channels by mCherry-CaMKII1–274, K42M-KRastail. Same format as (c). Data are means±s.e.m, n=5 for each point. *P<0.05 compared with control data by two-tailed Student’s paired t-test.
We next considered whether we could convert Ca2+-calmodulin-dependent protein kinase II (CaMKII) into a CCB. CaMKII has been found to constitutively bind CaV1.2 (ref. 29) and CaV2.1 (ref. 30) channels, respectively. CaMKII bound to CaV1.2 C terminus was found necessary for Ca2+-dependent facilitation of ICa (ref. 29). Further, CaMKII binding to CaV2.1 C terminus slows channel inactivation kinetics and regulates short-term synaptic plasticity in neurons30. CaMKII holo-enzyme is a multimer of 12 monomeric subunits35. Each monomer (475 amino acids) has three distinct domains: an N-terminal catalytic domain (residues 1–274) that mediates kinase activity, a central regulatory domain (residues 275–314) that exerts basal auto-inhibitory control of the kinase domain and an association domain (residues 315–475) that mediates subunit assembly. The catalytic domain of CaMKII mediates binding to CaV1.2 channels29. To assess the possibility of converting CaMKII into a CCB we fused either C1PKCγ or the polybasic membrane-targeting tail from K-Ras to the C terminus of CaMKII catalytic domain (residues 1–274). We also introduced a K42M point mutation that renders the kinase catalytically dead35. CaMKII[1–274,K42M]-C1PKC inducibly translocated to the membrane (Fig. 3f) and inhibited CaV2.2 and CaV1.2 channels in response to PdBu (Fig. 3g). Surprisingly, CaV2.1 channels were not inhibited by membrane-translocated CaMKII[1–274,K42M]-C1PKC (Fig. 3g, middle). By contrast, CaMKII[1–274,K42M]-KRastail was constitutively associated with the plasma membrane (Fig. 3h), and caused a deep inhibition of all three CaV channels (Fig. 3i). Beyond providing an additional proof of the principle for generating novel genetically encoded CCBs, these results suggest that recombinant CaV2.2 channels may also bind CaMKII. The discrepancy in CaV2.1 sensitivity to CaMKII[1–274,K42M]-C1PKC and CaMKII[1–274,K42M]-KRastail suggests that inhibition by the CaMKII-based inhibitor in this channel may be kinetically slow such that it is only apparent with the constitutive CCB.
Finally, we explored the feasibility of using CaM to create a genetically encoded CCB. CaM is known to bind CaV1 and CaV2 channels and mediates their regulation by Ca2+ ions. We generated mCherry-CaM-C1PKC, which displayed basal cytosolic localization but was translocated to the plasma membrane with PdBu (Fig. 4a). As the endogenous CaM concentration is relatively high, the efficacy of CaM-based CCBs would be expected to depend critically on how effectively they displace endogenous CaM from the channels. We used Ca2+-dependent inactivation (CDI) of CaV1.2 channels as a biosensor to gain insights into how effectively C1PKC-tagged wt and mutant CaM displaced endogenous CaM. When co-expressed with mCherry-CaM-C1PKC, recombinant CaV1.2 channels (α1C+β2a) displayed Ba2+ currents that showed a slow monotonic voltage-dependent inactivation (Fig. 4b). With Ca2+ as charge carrier, the same channels exhibited a fast and deep decrease in current amplitude with the kinetic signature of CDI (Fig. 4b). When co-expressed with a mutant mCherry-CaM1234-C1PKC with all four EF hands mutated so they no longer bind Ca2+, CaV1.2 channels displayed Ca2+ currents in which CDI was virtually eliminated (Fig. 4b). This result indicates that the over-expressed mCherry-CaM1234-C1PKC effectively out-competes endogenous CaM for binding to CaV1.2, and further demonstrates that the tags do not interfere with CaM binding to CaV channels. We found CaV2.2 channels co-expressed with mCherry-CaM1234-C1PKC were rapidly inhibited by PdBu (Fig. 4c). Surprisingly, both CaV2.1 and CaV1.2 co-expressed with mCherry-CaM1234-C1PKC were unaffected by PdBu (Fig. 4c). As CaM binds to all three channels32,33,36,37, these results suggest that the mere existence of a binding site for a cytosolic protein on the channel may not be sufficient to generate an inducible CCB in all cases. Another possibility is that potential inhibition of CaV2.1 and CaV1.2 induced by membrane-targeting mCherry-CaM1234-C1PKC is kinetically slow such that it does not occur during the 5–10 min time course of our electrophysiological assay.

(a) Confocal image showing PdBu-induced translocation of mCherry-CaM-C1PKC from cytosol to the plasma membrane in a transfected HEK 293 cell. Scale bar, 5 μm. (b) The C1PKC tag does not affect the functional interaction of CaM or CaM1234 with CaV1.2. Scale bar, 0.5 nA, 10 ms. (c) Diary plots and population bar charts showing PdBu-induced effects of CaV2.2, CaV2.1 and CaV1.2 channels co-expressed with mCherry-CaM-C1PKC. Data are means±s.e.m.
Overall, these data demonstrate that diverse intracellular proteins interacting with CaV1/CaV2 channels can be converted into constitutive or inducible CCBs with distinctive potency and/or selectivity. We have termed this general method Channel Inactivation induced by Membrane-tethering of an associated Protein (ChIMP). The acronym is apropos given the imagery of closing a channel pore by the induced ‘swinging’ of an associated protein from the cytoplasm to the plasma membrane (Fig. 2a).
Effectiveness of 14-3-3-based CCB on native CaV channels
The results to this point have tested the efficacy of genetically encoded CCBs on recombinant CaV channels reconstituted in HEK 293 cells. As native CaV channels are typically associated with macromolecular complexes and have a more complicated nano-enviroment than recombinant channels in heterologous cells, it was important to verify that the genetically engineered CCBs were effective against native CaV channels. We first examined the impact of mem-mCherry-14-3-3ε on CaV channels recorded from primary cultures of murine dorsal root ganglion (DRG) neurons. DRG neurons express multiple CaV1/CaV2 channel currents38. We used adenoviral vectors to express either mCherry-14-3-3 or mem-mCherry-14-3-3 (Fig. 5a) in cultured DRG neurons. Cells expressing mCherry-14-3-3 displayed an I–V relationship that was indistinguishable from that obtained with control uninfected neurons (Fig. 5b, left; Ipeak at 0 mV=57.2±9.9 pA/pF, n=6 for control neurons, Ipeak=62.0±19.6 pA/pF, n=6 for mCherry-14-3-3-expressing neurons). By contrast, expression of mem-mCherry-14-3-3ε in DRG neurons markedly suppressed endogenous IBa within 24 h of adenoviral infection (Fig. 5b, right; Ipeak=6.3±1.5 pA/pF, n=6 for mem-mCherry-14-3-3-expressing neurons, P<0.05 compared with control using unpaired t-test). We obtained similar results for nerve growth factor (NGF)-differentiated rat pheochromocytoma (PC12) cells (Fig. 5c), which contain CaV2.2 and CaV1.2 channels that trigger exocytosis39. These results demonstrate that genetically engineered CCBs developed according to the ChIMP principle are also effective against native CaV1/CaV2 channels despite their more elaborate nano-environment.

(a) Greyscale and fluorescence image of DRG neuron expressing mem-mCherry-14-3-3. Scale bar, 5 μm. (b) Population I–V curves in DRG neurons. Uninfected neurons (black triangles) compared with neurons expressing either mCherry-14-3-3 (left) or mem-mCherry-14-3-3 (right), n=6 for each point. (c) Population I–V curves in differentiated PC12 cells, same format as (b), n=5 for each point. *P<0.05 compared with control data by two-tailed Student’s paired t-test.
Potential use of ChIMP to discover small-molecule CCBs
The results so far suggest the general principle that the discovery process for new CCBs may be decomposed into two parts: first, finding a molecule that binds an appropriate α1-subunit cytoplasmic region (a ‘handle’) and second, a transduction step involving the anchoring of the ‘handle’ molecule to the plasma membrane. We next considered two ancillary issues. First, whether the ‘handle’ could be a small molecule rather than a protein as we have so far demonstrated. If so, the ChIMP method could potentially be extended to small-molecule drug discovery. Second, whether it would be possible to develop a method to systematically identify which areas in α1 cytoplasmic regions are appropriate target-binding sites that are permissive for regulated closure of the channel pore. To concurrently address these two issues, we adapted a heterodimerization strategy that relies on the ability of the small-molecule rapamycin to simultaneously bind two proteins, FK506-binding protein (FKBP) and a fragment of the mammalian target of rapamycin (FRB), respectively40. We inserted FKBP (one insert per channel) at four different positions within CaV1.2 α1C subunit intracellular regions (N terminus, I–II loop, and at proximal and distal positions in the C terminus) (Fig. 6a). All these constructs expressed currents when co-expressed with β2a and a constitutively membrane-targeted FRB (LDR) in HEK 293 cells18,41. Rapamycin (1 μM) caused a rapid inhibition of IBa in channels with FKBP inserted into the C terminus (Fig. 6d,e). By contrast, channels with FKBP inserted at the N terminus and in the distal I–II loop, respectively, did not display rapamycin-induced decrease in current (Fig. 6b,c). Neither of the two C terminus FKBP-fused channels responded to rapamycin in the absence of LDR (Supplementary Fig. S7). These findings offer a critical proof-of-concept that the ChIMP approach can potentially be used to discover new CCBs by using high-throughput screening to find molecules that bind appropriate ‘handle sites’ in CaV α1-subunits, and conjugating them to a membrane-targeting module. A major challenge that needs to be overcome to realize this possibility is developing sensitive high-throughput screens to identify small molecules that bind intracellular domains of CaV channels. One possibility is to use purified tagged CaV α1 intracellular loops to probe small-molecule microarrays42,43.

(a) Insertion of FKBP into selected regions of α1C intracellular loops/termini. (b–e) Impact of rapamycin on IBa from distinct FKBP-fused α1C constructs co-transfected with LDR. Scale bar, 400 pA, 10 ms.
Discussion
In this work we report the discovery that diverse proteins/molecules that bind distinct sites in intracellular loops of CaV1/CaV2 channels can be used as ‘handles’ to inhibit ICa through their controlled anchoring to the plasma membrane. This new insight paves the way for developing customized CCBs with selectivity for distinct CaV1/CaV2 channels based on the identity of the pore-forming α1-subunit, the auxiliary CaVβ or other associated cytoplasmic proteins. Genetically encoded CCBs are potentially desirable because they can be expressed in a geographically restricted fashion thereby eliminating off-target effects that may confound small-molecule drug therapy2,10. Furthermore, genetically encoded CCBs can be engineered to block molecularly identical CaV channels with sub-cellular specificity. For example, a caveolae-targeted Rem has been shown to selectively inhibit caveolae-localized CaV1.2 channels in heart cells while sparing dyadic CaV1.2 channels that trigger muscle contraction14. As caveolae-localized CaV1.2 channels are hypothesized to selectively signal to pathological cardiac hypertrophy, it is proposed that caveolae-specific CaV1.2 channel inhibitors could be an effective therapy for adverse remodelling of the heart14. RGK proteins have so far been used in proof-of-concept experiments to demonstrate the utility of genetically encoded CCBs10,14. However, the potential clinical use of RGKs is complicated by their broad biological effects, diverse binding partners and lack of specificity and controllability11. This work introduces ChIMP as a generalized method for developing novel genetically encoded CCBs with distinct potency, selectivity and kinetics. A caveat for the work is that the CCBs we have generated so far are derived from naturally occurring proteins that also have their own specific functions and binding partners in cells. Nevertheless, the general insights obtained from developing these proteins can now be potentially coupled with new technologies for evolving protein molecules that bind to target sites with high specificity, such as DARPins44 and intrabodies45, to develop highly selective CaV1/CaV2 channel blockers. Another aspect of the ChIMP technology that can be greatly improved relates to the method for inducing membrane targeting of the ‘handle’ protein. Here we used PdBu and rapamycin-mediated heterodimerization as convenient tools to demonstrate proof-of-concept of the ChIMP method. However, PdBu activates endogenous protein kinase C, and rapamycin associates with endogenous FKBP and mammalian target of rapamycin (mTOR), a protein kinase involved in cell proliferation, growth and survival. Hence, these two agents may be inappropriate for potential in vivo applications. Recently, several genetically encoded dimerizers based on plant photoreceptors that permit light-regulated, reversible protein heterodimerization have been developed46,47,48. Marriage of light-regulated heterodimerization with ChIMP could provide a powerful general method for optogenetic control of CaV channels and is an exciting prospect for future experiments.
We previously reported that Rem, which normally constitutively targets to the plasma membrane and inhibits CaV channels, could be converted into a small-molecule-regulated inducible CCB by dynamically regulating its association with the plasma membrane18,19. However, it was unknown how targeting Rem to the membrane caused CaV channel inhibition. This work supports a model where membrane-targeted Rem uses CaVβ as a bridge to alter the conformation of the I–II loop in a manner that closes the channel. This is an important new insight into the mechanism of action of RGKs, particularly in light of data that have raised questions about a role for RGK binding to CaVβ in the mechanism for CaV channel inhibition49. More generally, regulation of channel gating by induced conformational changes in intracellular domains is a rather common phenomenon that occurs in many different ion channels. For example, cyclic nucleotides and Ca2+ control the opening of cyclic nucleotide-gated (CNG) and large conductance K+ (BK) channels, respectively, by binding to channel intracellular domains50,51. Moreover, engagement of cytoplamic domains with the plasma membrane through interaction with phosphoinositide lipids regulates the gating of ion channels such as the inward rectifier K+ channel Kir2.2 (ref. 52) and TRPV1 (ref. 53). From this perspective, the ChIMP approach could provide important insights into how distinct intracellular domains link to channel gating in different ion channels.
The major conceptual advance in this study is that diverse cytosolic proteins or small molecules that bind distinct sites in intracellular loops of CaV1/CaV2 channels can likewise be converted to constitutive or inducible CaV channel inhibitors according to the mode of their anchoring to the plasma membrane. This is a non-trivial advance because it suggests a general method for developing novel genetically encoded blockers for many ion channel types. Ion channels are ubiquitous and essential to the biology of all cell types, and their dysfunction underlies many human diseases54. Selective ion channel modulators are highly sought after as therapeutics and research tools. However, there is a lack of specific modulators for many ion channel species. The ChIMP strategy may potentially be applied to develop novel blockers for a broad cohort of ion channels.
A corollary benefit of the ChIMP approach is its potential to provide a robust functional readout as to whether individual proteins directly interact with intracellular domains of recombinant CaV1/CaV2 channels. In our study, we discovered for the first time that recombinant CaV1.2 binds 14-3-3 and that CaV2.2 binds CaMKII. A proteomic study of the CaV2 channel nano-environment in mammalian brain indicates these channels are associated with a protein network gathered from a pool of ~200 proteins with distinct abundance and preference for CaV2.1–CaV2.3 subtypes55. Our studies suggest how CaV1/CaV2 channels may be used as a biosensor to validate some of these putative protein interactions.
Methods
cDNA cloning
To generate fluorescent-protein-tagged constructs, cyan or yellow fluorescent protein (CFP or YFP) was amplified using PCR and cloned into pcDNA4.1 (Invitrogen) using KpnI and BamHI sites. CFP–Rem1–265–C1PKCγ and CFP–Rem1–250–C1PKCγ were generated by using overlap extension PCR to fuse residues 26–89 of mouse PKCγ to the C terminus of Rem1–265 and Rem1–250, respectively. The fusion product was subsequently cloned downstream of CFP using BamHI and EcoRI sites. To create C1PKCγ-Rem78–265–CFP, C1PKCγ was cloned into pcDNA4.1 (Invitrogen) using KpnI and BamHI sites. Rem78–265 and CFP were subsequently amplified and cloned downstream of C1PKCγ using BamHI/EcoRI and EcoRI/XbaI sites, respectively. C1PKCγ-YFP-Rem1–265 was produced by first using overlap extension PCR to fuse C1PKCγ to the N terminus of YFP. The resulting fusion product was cloned upstream of Rem1–265 using KpnI and BamHI sites. CFP-β3-C1PKCγ was generated by using overlap extension PCR to fuse C1PKCγ to the C terminus of β3. The fusion product was then cloned downstream of CFP using BamHI and EcoRI sites. To generate C1PKCγ-mcherry-14-3-3, we used overlap extension PCR to fuse C1PKCγ to the N terminus of mCherry. The fusion product was cloned into pcDNA4.1 (Invitrogen) using KpnI and BamHI sites. 14-3-3 was PCR amplified and cloned downstream of mcherry using BamHI and XhoI sites. To create mcherry-CaMKII1–274K42M-C1PKCγ, we used overlap extension PCR to fuse C1PKCγ to the C terminus of CaMKII1–274. The fusion product was cloned into pcDNA4.1 (Invitrogen) using BamHI and XhoI sites sites. mcherry was PCR-amplified and cloned upstream using KpnI and BamHI sites. Point mutation in mcherry-CaMKII1–274K42M-C1PKCγ was introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). To generate FKBP-fused α1C constructs, we used overlap extension PCR to fuse YFP to the C terminus of α1C. The fusion product was cloned into pcDNA3.1 (Invitrogen) using KpnI and XbaI sites. FKBP was inserted into distinct regions of α1C intracellular loops using In-fusion Cloning Kit (Clontech). All PCR products were verified by sequencing.
Cell culture and transfection
Low-passage-number HEK 293 cells were maintained in DMEM supplemented with 10% FBS and 100 μg ml−1 penicillin–streptomycin. For electrophysiology and flow cytometry experiments, HEK 293 cells cultured in 6-cm tissue culture dishes were transiently transfected with CaV1/CaV2 α1 (6 μg), β3 (6 μg), T antigen (2 μg) and the appropriate GEMIICC construct (4 μg), using the calcium phosphate precipitation method. Cells were washed with PBS 5–8 h after transfection and maintained in supplemented DMEM. For confocal microscopy experiments, transfected HEK 293 cells were replated onto fibronectin-coated culture dishes with No. 0 glass coverslip bottoms (MaTek). For electrophysiology experiments cells were replated onto fibronectin-coated glass coverslips 24 h after transfection.
Murine dorsal root ganglion (DRG) neurons were kindly provided by the laboratory of Dr Joachim Scholz (Columbia University). DRG neurons were maintained in 96.5 ml Neurobasal A medium supplemented with 2 ml B-27, 100 μg ml−1 Pen/strep, 0.5 ml L-glutamine, 50 ng ml−1 NGF, 2 ng ml−1 GDNF and 10 μM Ara-C. For electrophysiology experiments, DRG neurons cultured in 2-cm tissue culture dishes were infected with the appropriate adenovirus. Undifferentiated PC12 cells were maintained in RPMI supplemented with 10% horse serum, 5% FBS and 100 μg ml−1 penicillin–streptomycin. Differentiated PC12 cells were maintained in RPMI supplemented with 1% horse serum. NGF (50 ng ml−1) was added to media just prior to use. For electrophysiology experiments, PC12 cells cultured in 6-cm tissue culture dishes were infected with the appropriate adenovirus.
Electrophysiology
Whole-cell recordings of HEK cells were conducted 48–72 h after transfection using an EPC—8 or EPC—10 patch clamp amplifier (HEKA Electronics) controlled by PULSE software (HEKA). Micropipettes were fashioned from 1.5-mm thin-walled glass with filament (WPI Instruments) and filled with internal solution containing (in mM): 135 caesium methanesulphonate (CsMeSO3), 5 CsCl, 5 EGTA, 1 MgCl2, 4 MgATP (added fresh) and 10 HEPES (pH 7.3). Series resistance was typically between 1.5–2 MΩ. There was no electronic series resistance compensation. External solution contained (in mM): 140 tetraethylammonium—MeSO3, 5 BaCl2, and 10 HEPES (pH 7.3). Whole-cell I–V curves were generated from a family of step depolarizations (−40 to +100 mV from a holding potential of −90 mV). Currents were sampled at 25 kHz and filtered at 5 or 10 kHz. Traces were acquired at a repetition interval of 6 s. Leak and capacitive currents were subtracted using a P/8 protocol.
Whole-cell recordings of DRG and PC12 cells were conducted 24–48 h after infection. HEK cell internal solution was used for both DRG and PC12 cells. HEK cell external solution was used for PC12 cells. HEK cell external solution with 0.5 μM TTX was used for DRG neurons.
Confocal microscopy
Static images of CFP–Rem1–265-C1PKCγ, CFP-β3-C1PKCγ, and mcherry-C1PKCγ-14-3-3 constructs were observed using a Leica TCS SPL AOBS MP Confocal microscope system and a × 40 oil objective (HCX PL APO 1.25–0.75 NA). HEK 293 cells expressing CFP, YFP and mCherry fusion proteins were imaged using the 458-, 514- and 543-nm Argon laser line, respectively, for excitation.
Data and statistical analyses
Data were analysed off-line using PulseFit (HEKA), Microsoft Excel and Origin software. Statistical analyses were performed in Origin using built-in functions. Statistically significant differences between means (P<0.05) were determined using Student’s t-test for comparisons between two groups or one-way ANOVA followed by pairwise means comparisons using Bonferroni test for multiple groups. Data are presented as means±s.e.m.
Additional information
How to cite this article: Yang, T. et al. Bio-inspired voltage-dependent calcium channel blockers. Nat. Commun. 4:2540 doi: 10.1038/ncomms3540 (2013).
References
Catterall, W. A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 (2000).
Xu, X. & Colecraft, H. M. Engineering proteins for custom inhibition of Ca(V) channels. Physiology (Bethesda) 24, 210–218 (2009).
Anekonda, T. S. & Quinn, J. F. Calcium channel blocking as a therapeutic strategy for Alzheimer's disease: the case for isradipine. Biochim. Biophys. Acta 1812, 1584–1590 2011.
Kochegarov, A. A. Pharmacological modulators of voltage-gated calcium channels and their therapeutical application. Cell Calcium 33, 145–162 (2003).
Simuni, T. et al. Tolerability of isradipine in early Parkinson's disease: a pilot dose escalation study. Mov. Disord. 25, 2863–2866 (2010).
Triggle, D. J. Calcium channel antagonists: clinical uses—past, present and future. Biochem. Pharmacol. 74, 1–9 (2007).
Striessnig, J. et al. Structural basis of drug binding to L Ca2+ channels. Trends Pharmacol. Sci. 19, 108–115 (1998).
Uchitel, O. D. Toxins affecting calcium channels in neurons. Toxicon 35, 1161–1191 (1997).
Zuccotti, A. et al. Structural and functional differences between L-type calcium channels: crucial issues for future selective targeting. Trends Pharmacol. Sci. 32, 366–375 (2011).
Murata, M., Cingolani, E., McDonald, A. D., Donahue, J. K. & Marban, E. Creation of a genetic calcium channel blocker by targeted gem gene transfer in the heart. Circ. Res. 95, 398–405 (2004).
Yang, T. & Colecraft, H. M. Regulation of voltage-dependent calcium channels by RGK proteins. Biochim. Biophys. Acta 1828, 1644–1654 2012.
Beguin, P. et al. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411, 701–706 (2001).
Finlin, B. S., Crump, S. M., Satin, J. & Andres, D. A. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. Proc. Natl Acad. Sci. USA 100, 14469–14474 (2003).
Makarewich, C. A. et al. A caveolae-targeted L-type Ca(2)+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ. Res. 110, 669–674 (2012).
Correll, R. N., Pang, C., Niedowicz, D. M., Finlin, B. S. & Andres, D. A. The RGK family of GTP-binding proteins: regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell Signal. 20, 292–300 (2008).
Flynn, R. & Zamponi, G. W. Regulation of calcium channels by RGK proteins. Channels (Austin) 4, 434–439 (2010).
Yang, T., Puckerin, A. & Colecraft, H. M. Distinct RGK GTPases differentially Use alpha(1)- and auxiliary beta-binding-dependent mechanisms to inhibit Ca(V)1.2/Ca(V)2.2 channels. PLoS One 7, e37079 (2012).
Yang, T., Suhail, Y., Dalton, S., Kernan, T. & Colecraft, H. M. Genetically encoded molecules for inducibly inactivating CaV channels. Nat. Chem. Biol. 3, 795–804 (2007).
Yang, T., Xu, X., Kernan, T., Wu, V. & Colecraft, H. M. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J. Physiol. 588, 1665–1681 (2010).
Buraei, Z. & Yang, J. The {beta} subunit of voltage-gated Ca2+ channels. Physiol. Rev. 90, 1461–1506 (2010).
Chen, Y. H. et al. Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680 (2004).
Opatowsky, Y., Chen, C. C., Campbell, K. P. & Hirsch, J. A. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399 (2004).
Van Petegem, F., Clark, K. A., Chatelain, F. C. & Minor, D. L. Jr. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675 (2004).
Pragnell, M. et al. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature 368, 67–70 (1994).
Takahashi, S. X., Mittman, S. & Colecraft, H. M. Distinctive modulatory effects of five human auxiliary beta 2 subunit splice variants on L-type calcium channel gating. Biophys. J. 84, 3007–3021 (2003).
Chien, A. J. et al. Roles of a membrane-localized beta subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem 270, 30036–30044 (1995).
Restituito, S. et al. The [beta]2a subunit is a molecular groom for the Ca2+ channel inactivation gate. J. Neurosci. 20, 9046–9052 (2000).
Suh, B. C., Kim, D. I., Falkenburger, B. H. & Hille, B. Membrane-localized beta-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl Acad. Sci. USA 109, 3161–3166 (2012).
Hudmon, A. et al. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547 (2005).
Jiang, X. et al. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc. Natl Acad. Sci. USA 105, 341–346 (2008).
Li, Y., Wu, Y. & Zhou, Y. Modulation of inactivation properties of CaV2.2 channels by 14-3-3 proteins. Neuron 51, 755–771 (2006).
Peterson, B. Z., DeMaria, C. D., Adelman, J. P. & Yue, D. T. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 22, 549–558 (1999).
Lee, A. et al. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159 (1999).
Zuhlke, R. D., Pitt, G. S., Deisseroth, K., Tsien, R. W. & Reuter, H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399, 159–162 (1999).
Chao, L. H. et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell 146, 732–745 (2011).
Liang, H. et al. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39, 951–960 (2003).
Van Petegem, F., Chatelain, F. C. & Minor, D. L. Jr. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat. Struct. Mol. Biol. 12, 1108–1115 (2005).
Scroggs, R. S. & Fox, A. P. Multiple Ca2+ currents elicited by action potential waveforms in acutely isolated adult rat dorsal root ganglion neurons. J. Neurosci. 12, 1789–1801 (1992).
Taylor, S. C. & Peers, C. Store-operated Ca2+ influx and voltage-gated Ca2+ channels coupled to exocytosis in pheochromocytoma (PC12) cells. J. Neurochem. 73, 874–880 (1999).
Crabtree, G. R. & Schreiber, S. L. Three-part inventions: intracellular signaling and induced proximity. Trends Biochem. Sci. 21, 418–422 (1996).
Inoue, T., Heo, W. D., Grimley, J. S., Wandless, T. J. & Meyer, T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 (2005).
Bradner, J. E. et al. A robust small-molecule microarray platform for screening cell lysates. Chem. Biol. 13, 493–504 (2006).
Noblin, D. J. et al. A HaloTag-based small molecule microarray screening methodology with increased sensitivity and multiplex capabilities. ACS Chem. Biol. 7, 2055–2063 (2012).
Boersma, Y. L. & Pluckthun, A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr. Opin. Biotechnol. 22, 849–857 (2011).
Zhou, C. & Przedborski, S. Intrabody and Parkinson’s disease. Biochim. Biophys. Acta 1792, 634–642 (2009).
Kennedy, M. J. et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975 (2010).
Levskaya, A., Weiner, O. D., Lim, W. A. & Voigt, C. A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009).
Yazawa, M., Sadaghiani, A. M., Hsueh, B. & Dolmetsch, R. E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 27, 941–945 (2009).
Fan, M., Buraei, Z., Luo, H. R., Levenson-Palmer, R. & Yang, J. Direct inhibition of P/Q-type voltage-gated Ca2+ channels by Gem does not require a direct Gem/Cavbeta interaction. Proc. Natl Acad. Sci. USA 107, 14887–14892 (2010).
Craven, K. B. & Zagotta, W. N. CNG and HCN channels: two peas, one pod. Annu. Rev. Physiol. 68, 375–401 (2006).
Salkoff, L., Butler, A., Ferreira, G., Santi, C. & Wei, A. High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931 (2006).
Hansen, S. B., Tao, X. & MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498 (2011).
Cao, E., Cordero-Morales, J. F., Liu, B., Qin, F. & Julius, D. TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77, 667–679 (2013).
Ashcroft, F. M. Ion Channels and Disease Academic Press (1999).
Muller, C. S. et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc. Natl Acad. Sci. USA 107, 14950–14957 (2010).
Acknowledgements
We thank Drs T. Meyer (Stanford University) and T. Inoue (Johns Hopkins University) for FRB construct (LDR), Drs Ademuyiwa Aromolaran and Prakash Subramanyam for comments on the manuscript, Dr David Yue (Johns Hopkins University) for discussions, Drs Llloyd Greene and Oren Levy (Columbia University) for PC12 cells, Dr Joachim Scholz (Columbia University) for DRG neurons, and Brian Soda for assistance with the ChIMP illustration artwork. This work was supported by NIH grants RO1 HL084332 and RO1 HL069911 (to H.M.C). L.L.H. was supported by a NIH postdoctoral training grant (T32 HL087745). H.M.C. is an Established Investigator of the American Heart Association.
Author information
Authors and Affiliations
Contributions
L.-L.H. generated the plasmid constructs used in Figs 1 and 2, designed experiments, performed electrophysiological experiments and analyses for Figs 1 and 2; T.T.Y. performed all electrophysiological experiments and analyses for Figs 3, 4, 5 and 6, performed some electrophysiological experiments and analyses for Fig. 2, designed experiments, made figures and helped write the paper; M.C. generated the constructs used in Figs 3, 4 and 5; K.F. generated the constructs used in Fig. 6; H.M.C. obtained funding, thought of the concept, analysed the data, made figures and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S7 (PDF 832 kb)
Rights and permissions
About this article
Cite this article
Yang, T., He, LL., Chen, M. et al. Bio-inspired voltage-dependent calcium channel blockers. Nat Commun 4, 2540 (2013). https://doi.org/10.1038/ncomms3540
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms3540
This article is cited by
-
Investigation and Restoration of BEST1 Activity in Patient-derived RPEs with Dominant Mutations
Scientific Reports (2019)
-
Dual Ca2+-dependent gates in human Bestrophin1 underlie disease-causing mechanisms of gain-of-function mutations
Communications Biology (2019)
-
ATP activates bestrophin ion channels through direct interaction
Nature Communications (2018)
-
Functional assessment of three Rem residues identified as critical for interactions with Ca2+ channel β subunits
Pflügers Archiv - European Journal of Physiology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
