
Abstract
Distinguishing insulators by the dominant type of interaction is a central problem in condensed matter physics. Basic models include the Bloch-Wilson and the Peierls insulator due to electron–lattice interactions, the Mott and the excitonic insulator caused by electron–electron interactions, and the Anderson insulator arising from electron–impurity interactions. In real materials, however, all the interactions are simultaneously present so that classification is often not straightforward. Here, we show that time- and angle-resolved photoemission spectroscopy can directly measure the melting times of electronic order parameters and thus identify—via systematic temporal discrimination of elementary electronic and structural processes—the dominant interaction. Specifically, we resolve the debates about the nature of two peculiar charge-density-wave states in the family of transition-metal dichalcogenides, and show that Rb intercalated 1T-TaS2 is a Peierls insulator and that the ultrafast response of 1T-TiSe2 is highly suggestive of an excitonic insulator.
Similar content being viewed by others


Introduction
Metal-insulator transitions have a prominent role in condensed matter physics, not only as a source of spectacular resistivity changes over many orders of magnitude that may form the basis of electronic devices, but also particularly as a major intellectual challenge driving the development of theoretical and experimental methods1,2,3,4,5. The active interest in the field is largely due to the fact that one is confronted with a many-body problem. As any metal-insulator transition is driven by a variety of interactions between the many electronic and structural degrees of freedom constituting a solid, the phenomenon represents an excellent test of our understanding of complex systems4.
In systems dominated by electron–electron and electron–phonon interactions, there are three distinct models of insulators, all connected with the emergence of long-range charge order and a gap in the electronic structure at the Fermi energy EF (Fig. 1)2,5. The first model is the Mott insulator, resulting from the intra-atomic Coulomb interaction. Mott insulators can arise from metals with a flat band straddling EF. They are characterized by the complete localization of the valence electrons at the atomic sites and by the opening of a density-of-states gap at EF (Fig. 1a). The second model is the excitonic insulator, arising from the unscreened electron–hole interaction. An excitonic instability due to the spontaneous formation of excitons can occur in a semimetal with a small overlap or a semiconductor with a small gap and may lead to a charge-density wave (CDW). The CDW phase is accompanied by a small, 'accidental' lattice distortion due to finite electron–phonon coupling, and is stabilized by a gap at EF due to an avoided crossing of the valence and conduction bands (Fig. 1b). The third model is the Peierls insulator, resulting from electron–phonon interactions. In a Peierls instability, the formation of a CDW and a periodic lattice distortion go hand in hand. The wavelength and symmetry of the periodic modulations fit to the Fermi surface such that occupied electronic states near EF are lowered and the resulting gain in electronic band energy is larger than the cost in Coulomb and lattice energy (Fig. 1c).

(a) Mott insulator. (b) Excitonic insulator. (c) Peierls insulator. For each case, the characteristic signatures of the insulating state (solid lines, coloured shading) following the transition from a metallic state (dashed lines, grey shading) are illustrated: in real space, the periodic modulation of the valence-electron density and, if applicable, of the atomic positions, and in energy-momentum space, the opening of an energy gap (Δ). (d) Corresponding timescales of the responses to impulsive near-infrared excitation, as derived from the materials investigated in the present work, and their assignment to elementary model-specific processes.
Classification according to the above scheme, however, is often not straightforward because in real complex materials, electron–electron and electron–phonon interactions are often simultaneously strong and the dominant interaction is not easily identified. An important part of the problem is that the characteristic signature of the resulting insulating state, the gap in the electronic spectrum as observed in (time-integrated) spectroscopy, may look the same in all the three cases.
The central hypothesis of the present work is that complex insulators can be classified more reliably in the time domain—by time- and momentum-resolved pump-probe spectroscopy—on the basis of the gap melting times. The underlying idea is that the length and excitation density dependence of the melting time should directly reflect an elementary interaction-dependent process specific to each class of insulator (Fig. 1d). This hypothesis is here tested using a set of transition-metal dichalcogenides, layered materials that continue to provide a fertile field for disentangling the combined effects of electron–phonon and electron–electron interactions, in pristine compounds6 as well as in intercalation complexes7. Two prominent examples are 1T-TaS2 and 1T-TiSe2. In these two compounds, the electronic structures8,9,10,11,12,13,14,15 and ultrafast spectral responses16,17,18,19 indicate the presence of significant electron correlation effects. In both compounds, electron–electron interactions may in fact have an important role in CDW formation12,13,14,18,19,20 and in the emergence of superconductivity from CDW states upon intercalation21 or under pressure22,23.
Results
Systems
The momentum-space symmetries of the three arguably most intriguing CDW phases of 1T-TaS2 and 1T-TiSe2, are shown in Fig. 2, together with the unreconstructed schematic Fermi surfaces and the reconstructed band structures as seen by (time-integrated) angle-resolved photoemission spectroscopy (ARPES).

Reconstructed (green lines) and original (black lines) projected Brillouin zones (top row) and measured band dispersions along the  high-symmetry path (bottom row) for (a,b) the
high-symmetry path (bottom row) for (a,b) the  phase of 1T-TaS2 (c,d) the p(2×2×2) phase of 1T-TiSe2 and (e,f) the
phase of 1T-TaS2 (c,d) the p(2×2×2) phase of 1T-TiSe2 and (e,f) the  phase of Rb:1T-TaS2. In (a,c,e), unreconstructed schematic Fermi surfaces (circle and ellipses), wavevectors of the CDW (arrows) and high-symmetry points of the original Brillouin zones (dots) are indicated. For illustrative purposes, unreconstructed 1T-TiSe2 is assumed to be semimetallic. In (b,d,f), temperatures and photon energies of the ARPES measurements are indicated. Photoemission intensity is represented in a false-colour scale. The arrows mark characteristic energy gaps in the spectra.
phase of Rb:1T-TaS2. In (a,c,e), unreconstructed schematic Fermi surfaces (circle and ellipses), wavevectors of the CDW (arrows) and high-symmetry points of the original Brillouin zones (dots) are indicated. For illustrative purposes, unreconstructed 1T-TiSe2 is assumed to be semimetallic. In (b,d,f), temperatures and photon energies of the ARPES measurements are indicated. Photoemission intensity is represented in a false-colour scale. The arrows mark characteristic energy gaps in the spectra.
The  reconstruction of pristine 1T-TaS2 (Fig. 2a), which occurs at temperatures below 180–200 K (depending on cooling or heating), is rather well understood as a combined Peierls–Mott insulating phase8,10,24: the strong periodic lattice distortion, which is coupled to the CDW, splits the occupied part of the lowest-lying Ta 5d band into three subband manifolds so that the uppermost, half-filled manifold becomes susceptible to a Mott-Hubbard-type transition creating a deep pseudogap at EF. The spectroscopic signatures of this scenario are remarkably strong and momentum selective. In the ARPES spectral weight distribution depicted in Fig. 2b, the Mott-Hubbard gap (taken here as the distance between EF and the lower Hubbard band) has a size of about 120 meV and is best visible near the
reconstruction of pristine 1T-TaS2 (Fig. 2a), which occurs at temperatures below 180–200 K (depending on cooling or heating), is rather well understood as a combined Peierls–Mott insulating phase8,10,24: the strong periodic lattice distortion, which is coupled to the CDW, splits the occupied part of the lowest-lying Ta 5d band into three subband manifolds so that the uppermost, half-filled manifold becomes susceptible to a Mott-Hubbard-type transition creating a deep pseudogap at EF. The spectroscopic signatures of this scenario are remarkably strong and momentum selective. In the ARPES spectral weight distribution depicted in Fig. 2b, the Mott-Hubbard gap (taken here as the distance between EF and the lower Hubbard band) has a size of about 120 meV and is best visible near the  point8. The CDW reconstruction, on the other hand, seems most pronounced in the clear subband splittings along
point8. The CDW reconstruction, on the other hand, seems most pronounced in the clear subband splittings along  , the strong band backfolding along
, the strong band backfolding along  and the large spectral gap of about 400 meV that opens at the edge of the original Brillouin zone (midway between
and the large spectral gap of about 400 meV that opens at the edge of the original Brillouin zone (midway between  and
and  ). The latter feature may be understood as the projection of the original, nesting-induced gap of the high-temperature incommensurate CDW phase into the low-temperature commensurate
). The latter feature may be understood as the projection of the original, nesting-induced gap of the high-temperature incommensurate CDW phase into the low-temperature commensurate  phase15 (as indicated by the nesting vector in Fig. 2a).
phase15 (as indicated by the nesting vector in Fig. 2a).
Second, there is the conspicuous p(2 × 2 × 2) CDW phase (Fig. 2c) of pristine 1T-TiSe2, appearing below 200 K25, whose microscopic origin has remained elusive for more than three decades. On the one hand, quantitative microscopic explanations26 are difficult and deviate too much from experiment12,13,14. On the other hand, qualitative explanations in terms of an electron-lattice instability (variously called Peierls instability, band-type/indirect Jahn-Teller effect or antiferroelectric transition)12,25,27,28,29, a purely electronic phenomenon (an excitonic insulator instability)14,20,30, or a combination of the two scenarios13,31 are ambiguous as they all seem consistent with subsets of the extensive body of experimental and theoretical results. All proposed models explicitly involve the interaction between the Se 4p valence band maximum at  and the conduction band minimum at , the band extrema that are 'nested' by the wavevector of the CDW and mapped onto each other in the CDW phase (Fig. 2c). The spectral weight distribution in Fig. 2d shows that the ensuing direct band repulsion results in a gap of about 100 meV between the lowest-lying Ti 3d states and the strikingly intense Se 4p states folded to the original point. The controversy about the CDW mechanism is ultimately about the nature of this p–d interaction, about whether it is predominantly phononic or excitonic.
and the conduction band minimum at , the band extrema that are 'nested' by the wavevector of the CDW and mapped onto each other in the CDW phase (Fig. 2c). The spectral weight distribution in Fig. 2d shows that the ensuing direct band repulsion results in a gap of about 100 meV between the lowest-lying Ti 3d states and the strikingly intense Se 4p states folded to the original point. The controversy about the CDW mechanism is ultimately about the nature of this p–d interaction, about whether it is predominantly phononic or excitonic.
The third peculiar CDW phase occurs in intercalation complexes of 1T-TaS2 (ref. 32), in particular upon in situ Rb intercalation of pristine 1T-TaS2 (Rb:1T-TaS2) at room temperature33,34. The phase is characterized by a non-hexagonal  . CDW superlattice (Fig. 2e) and, in the measured spectral weight distribution, by a surprisingly large energy gap of about 400 meV in the Ta 5d states at EF around the
. CDW superlattice (Fig. 2e) and, in the measured spectral weight distribution, by a surprisingly large energy gap of about 400 meV in the Ta 5d states at EF around the  point (Fig. 2f). However, the nature of this gap is presently unclear: while the intercalation-induced change of CDW symmetry clearly points to a Peierls gap, the magnitude and weak momentum dependence of the gap, the absence of Fermi surface nesting (Fig. 2e), and the lack of substructure in the band dispersion rather suggest the gap to be of the Mott type33,34.
point (Fig. 2f). However, the nature of this gap is presently unclear: while the intercalation-induced change of CDW symmetry clearly points to a Peierls gap, the magnitude and weak momentum dependence of the gap, the absence of Fermi surface nesting (Fig. 2e), and the lack of substructure in the band dispersion rather suggest the gap to be of the Mott type33,34.
In summary, the three selected CDW phases confront us with three intriguing variations of the possible interplay of electron–phonon and electron–electron (electron–hole) interactions in the formation of a gapped ground state, in each case raising the intertwined questions about the nature of this ground state and the origin of the transition. In addition, the selection of materials allows for a systematic exploration of these questions because after all the unreconstructed systems exhibit the same basic structure and, despite their distinct band structures, a similar density of states near EF15. For pristine 1T-TaS2 the question seems indeed settled: the material is a Peierls-Mott insulator, in which the Peierls instability provides a sufficient condition for the Mott transition to occur. But for the other two systems the situation is unclear and the general classification problem reduces to the two specific questions: Is Rb:1T-TaS2 a Mott or a Peierls insulator and is 1T-TiSe2 a Peierls or an excitonic insulator?
Time-resolved ARPES
We now set out to answer these questions by time-resolved pump-probe ARPES16,17,18,19,35,36 using a high-harmonic-generation source37,38. The central idea is that the timescale on which electronic order in momentum space melts should provide a strong hint as to its predominant nature or origin. To verify this idea, we will establish a hierarchy of melting times for different types of spectral features in the three systems that shall allow us to identify the dominant interaction behind the gapped phases of Rb:1T-TaS2 and 1T-TiSe2, if we use the characteristic momentum-dependent spectral response of 1T-TaS216,17,18,39,40 as a reference. Specifically, we will compare and contrast the dynamics of the Ta 5d gap at  in Rb:1T-TaS2 (Fig. 2f) and the folded Se 4p spectral weight at in 1T-TiSe2 (Fig. 2d) with the dynamics of the Mott gap at
in Rb:1T-TaS2 (Fig. 2f) and the folded Se 4p spectral weight at in 1T-TiSe2 (Fig. 2d) with the dynamics of the Mott gap at  and the Peierls gap near in pristine 1T-TaS2 (Fig. 2b).
and the Peierls gap near in pristine 1T-TaS2 (Fig. 2b).
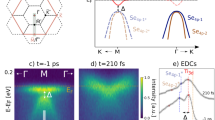
In our time-resolved ARPES experiments, to facilitate a systematic comparison and reveal any excitation-density dependence, the four 'spectroscopic order parameters' in the three different materials are quenched under identical excitation conditions for two different excitation densities. All samples are held at an equilibrium temperature of 110 K and are impulsively excited with 30 fs (FWHM), 790 nm near-infrared pulses at constant absorbed energy densities of 300 and 600 Jcm−3. At these excitation densities, the lattice temperatures in all three systems will transiently increase by 163±21 and 314±41 K, respectively, enough to drive pristine 1T-TaS2 and 1T-TiSe2 across thermal phase transitions (see Methods). Yet, the ARPES snapshots in Fig. 3a-d display qualitative electronic structure changes happening within 65–210 fs after excitation, which is much faster than the 0.35–4 ps it takes to transfer the excess energy in the electron system to the phonon bath16,17,19,39,40,41. These changes therefore do not reflect transitions to quasi-equilibrium states at elevated effective temperatures, although the band maps look fairly similar to temperature-dependent equilibrium ARPES data9,12,13,14.

(a–d) ARPES snapshots taken before optical pumping and at characteristic pump-probe delays: (a) at the  point of 1T-TaS2 (b) at the point of 1T-TiSe2 (c) at the
point of 1T-TaS2 (b) at the point of 1T-TiSe2 (c) at the  point of Rb:1T-TaS2 and (d) at the point of 1T-TaS2 (absorbed pump energy density: 300 Jcm−3; equilibrium temperature: 110 K). Solid lines in (b,d) are guides to the eye. Arrows in (a,c,d) mark energy gaps. (e–l) Time-dependent ARPES spectra at selected momenta for absorbed pump energy densities of (e–h) 300 Jcm−3 and (i–l) 600 Jcm−3: (e,i) at the
point of Rb:1T-TaS2 and (d) at the point of 1T-TaS2 (absorbed pump energy density: 300 Jcm−3; equilibrium temperature: 110 K). Solid lines in (b,d) are guides to the eye. Arrows in (a,c,d) mark energy gaps. (e–l) Time-dependent ARPES spectra at selected momenta for absorbed pump energy densities of (e–h) 300 Jcm−3 and (i–l) 600 Jcm−3: (e,i) at the  point of 1T-TaS2, (f,j) at the point of 1T-TiSe2, (g, k) at the
point of 1T-TaS2, (f,j) at the point of 1T-TiSe2, (g, k) at the  point of Rb:1T-TaS2, and (h, l) near the point of 1T-TaS2. The respective absorbed pump fluences are indicated. Arrows in (e–h) mark the pump-probe delays of the data shown in the right panels of (a–d). The momentum intervals over which the energy distribution curves have been integrated are [0,0.1] Å−1 in (e–g, i–k) and [0.2, 0.3] Å−1 in (h,l).
point of Rb:1T-TaS2, and (h, l) near the point of 1T-TaS2. The respective absorbed pump fluences are indicated. Arrows in (e–h) mark the pump-probe delays of the data shown in the right panels of (a–d). The momentum intervals over which the energy distribution curves have been integrated are [0,0.1] Å−1 in (e–g, i–k) and [0.2, 0.3] Å−1 in (h,l).
The most prominent spectral changes are, on the one hand, the (partial) collapses of the various energy gaps at EF in 1T-TaS2 (Fig. 3a,d) and in Rb:1T-TaS2 (Fig. 3c), essentially resulting from shifts of the topmost spectral features to higher energies, and, on the other hand, the suppression of the superstructure peak in 1T-TiSe2 at the point (Fig. 3b), as indicated by the transfer of spectral weight to electron states above EF, the narrowing of the spectrum at , and the apparent switch of the band dispersion from hole-like to electron-like19. We also note that despite the poorer energy resolution compared to the synchrotron data of Fig. 2, all four 'spectroscopic order parameters' are clearly resolved in Fig. 2a–d and that the impulsive near-infrared pumping does not degrade the resolution.
Figure 3e–l shows the corresponding diverse electronic structure dynamics at selected momenta for the two pump densities used, displaying temporal responses that vary significantly in their rise time, character of the decay and excitation density dependence. The initial closings of the Mott gap and the Peierls gap in the reference compound 1T-TaS2 are clearly seen as the fastest and slowest processes, respectively, with rise times of <50 and ~200 fs that seem to be independent of the excitation density (Fig. 3e,h,i,l). On longer timescales, the time-resolved ARPES spectra at the corresponding momenta are periodically modulated, as is particularly obvious for the lower excitation density (Fig. 3e,h). This oscillatory component in the spectral response is known to reflect the coherent amplitude-mode oscillations of the CDW which in 1T-TaS2 couple strongly to the electrons near EF16,18. At the higher excitation density, these oscillations are strongly damped and become anharmonic (Fig. 3i,l)42. Figure 3f,j,g,k shows that the melting of the superstructure peak at in 1T-TiSe2 and the closing of the energy gap at  in Rb:1T-TaS2 happen on intermediate timescales and that the oscillations in the spectral responses are completely suppressed, except for a barely visible oscillation of the leading valence-band edge in Rb:1T-TaS2 at the lower excitation density (Figs 3g and 4). For 1T-TiSe2, the absence of coherent amplitude-mode oscillations is consistent with time-resolved optical reflectivity results which show that these oscillations are strongly damped at the sample temperature and excitation densities used in our experiments41. A distinctive feature of the electronic structure dynamics of 1T-TiSe2 is that the melting time of the superstructure peak is dependent on the excitation density, the faster melting occurring at the higher density (see energy range between −0.6 and −0.2 eV between 0 and 100 fs in Fig. 3f,j)19.
in Rb:1T-TaS2 happen on intermediate timescales and that the oscillations in the spectral responses are completely suppressed, except for a barely visible oscillation of the leading valence-band edge in Rb:1T-TaS2 at the lower excitation density (Figs 3g and 4). For 1T-TiSe2, the absence of coherent amplitude-mode oscillations is consistent with time-resolved optical reflectivity results which show that these oscillations are strongly damped at the sample temperature and excitation densities used in our experiments41. A distinctive feature of the electronic structure dynamics of 1T-TiSe2 is that the melting time of the superstructure peak is dependent on the excitation density, the faster melting occurring at the higher density (see energy range between −0.6 and −0.2 eV between 0 and 100 fs in Fig. 3f,j)19.

Time dependence of the ARPES intensity in selected energy intervals of the transient energy distribution curves shown in Fig. 3g,h (absorbed pump energy density: 300 Jcm−3). The experimental data for 1T-TaS2 near the point (gold dots) and Rb:1T-TaS2 at the  point (violet dots) are compared with fits to a sine function with an exponentially decreasing amplitude (solid lines). The best-fit values for the oscillation period are indicated. Intensities are normalized to the average intensity I0 in the delay interval [−500,−50] fs; curves are offset for clarity. The energy intervals over which the intensity has been integrated are [−2,−1] eV for 1T-TaS2 and [−1.2,0] eV for Rb:1T-TaS2.
point (violet dots) are compared with fits to a sine function with an exponentially decreasing amplitude (solid lines). The best-fit values for the oscillation period are indicated. Intensities are normalized to the average intensity I0 in the delay interval [−500,−50] fs; curves are offset for clarity. The energy intervals over which the intensity has been integrated are [−2,−1] eV for 1T-TaS2 and [−1.2,0] eV for Rb:1T-TaS2.
The oscillatory components in the spectral responses of pristine and Rb intercalated 1T-TaS2 are analysed in Fig. 4 by plotting the ARPES intensities within selected energy intervals as a function of the pump-probe delay. The observed period of the amplitude-mode oscillation in pristine 1T-TaS2 is τamp=412±5 fs and agrees reasonably well with the bulk value of about 425 fs measured by time-resolved optical reflectivity at 120 K (ref. 43). The difference may be explained by the fact that our ARPES measurements, which employ extreme ultraviolet (XUV) radiation, are highly surface sensitive and probably only probe the stiffer surface amplitude mode16. In Rb:1T-TaS2, the amplitude of the oscillation is smaller, the damping stronger and τamp=234±6 fs shorter by almost a factor of two, all of which strongly indicate that the properties of the collective CDW excitations are significantly modified upon the Rb intercalation-induced transition to the  phase. However, what is important for the following quantitative analysis is that the experimentally determined values of τamp (or some fraction thereof) define an intrinsic timescale for the fastest possible structural response in the two systems16,17,35,36,40.
phase. However, what is important for the following quantitative analysis is that the experimentally determined values of τamp (or some fraction thereof) define an intrinsic timescale for the fastest possible structural response in the two systems16,17,35,36,40.
Figure 5 shows characteristic ARPES intensity transients, extracted from the data in Fig. 3e–l, that allow us to quantify how fast the various electronic order parameters are quenched (note that for better comparison the order-parameter transients of 1T-TiSe2 have been inverted). These transients also reveal part of the complex relaxation dynamics that will not be further investigated here. The detailed shape of the transients after the initial exponential rise generally depends on several intrinsic and extrinsic factors including the rate of electron-phonon thermalization, the presence and onset of coherent amplitude-mode oscillation, and the nature of the quasi-equilibrium state being reached, as well as the choice of the integration window and normalization procedure. The interplay of these factors can, for example, produce the plateau in the order-parameter transient of 1T-TaS2, to which we do not attribute a specific cause, and the seemingly slower relaxation dynamics of 1T-TiSe2, which does in fact happen on the picosecond timescale19. By contrast, because of its ultrashort timescale, the analysis of the melting dynamics is significantly more robust.

Time dependence of the ARPES intensity in selected energy intervals of the transient energy distribution curves shown in Fig. 3e–l for two absorbed pump energy densities of 300 Jcm−3 (left column) and 600 Jcm−3 (right column). The experimental data for 1T-TaS2 at the  point (blue dots, first row), 1T-TiSe2 at the point (green dots, second row), Rb:1T-TaS2 at the
point (blue dots, first row), 1T-TiSe2 at the point (green dots, second row), Rb:1T-TaS2 at the  point (violet dots, third row) and 1T-TaS2 near the point (gold dots, fourth row) are compared with fits to a model function described in the Methods section. The best-fit values for the melting time constants are indicated. Intensities are normalized to the average intensity I0 in the delay interval [−500,−50] fs; curves are offset for clarity. Note that for better comparison, the curves for 1T-TiSe2 have been inverted (they represent intensity losses in the chosen energy interval, whereas all other curves show intensity increases). The energy intervals over which the intensity has been integrated are [−0.12,0] eV at the
point (violet dots, third row) and 1T-TaS2 near the point (gold dots, fourth row) are compared with fits to a model function described in the Methods section. The best-fit values for the melting time constants are indicated. Intensities are normalized to the average intensity I0 in the delay interval [−500,−50] fs; curves are offset for clarity. Note that for better comparison, the curves for 1T-TiSe2 have been inverted (they represent intensity losses in the chosen energy interval, whereas all other curves show intensity increases). The energy intervals over which the intensity has been integrated are [−0.12,0] eV at the  point of 1T-TaS2, [−1.0,−0.4] eV at the point of 1T-TiSe2 and [−0.4,0] eV at the
point of 1T-TaS2, [−1.0,−0.4] eV at the point of 1T-TiSe2 and [−0.4,0] eV at the  point of Rb:1T-TaS2 and near the point of 1T-TaS2.
point of Rb:1T-TaS2 and near the point of 1T-TaS2.
Using a simple phenomenological model function (described in the Methods section), we can establish the envisioned hierarchy of melting time constants and link it to the timescales of elementary electronic and structural processes (as illustrated in Fig. 1d). The reference timescales are set by pristine 1T-TaS2 (top and bottom row of Fig. 5). The Mott gap collapses quasi-instantaneously within the duration of the pump pulse (30 fs FWHM), with a time constant of less than 20 fs, in agreement with the results of a recent time-resolved ARPES study18. This ultrashort timescale is direct evidence for the electronic origin of the gap; it is consistent with an electronic hopping time  , which sets the timescale for the response of the correlated electron system to sudden changes44,45 (here W=80–400 meV corresponds to the width of the split-off Ta 5d band that straddles EF10,16,11). The melting of the Peierls gap, by contrast, takes at least one order of magnitude longer, about 226±38 fs. This value corresponds to half an oscillation cycle of the amplitude mode τamp/2=206 fs, a connection which is immediately visible in Fig. 3h, l and a clear indication for the structural origin of the gap. The same fingerprint of Peierls physics is seen in the melting of the energy gap in Rb:1T-TaS2, though on a shorter timescale (third row of Fig. 5). The melting time constant is 126±20 fs and agrees well with the structural timescale, τamp/2=117 fs. The suppression of the superstructure peak in 1T-TiSe2, on the other hand, is qualitatively different and cannot be classified as easily (second row of Fig. 5). The process is slower than electron hopping (τhop<1 fs for W≳ 1eV (ref. 20)), but faster than atomic motion (τamp/2>160 fs at 110 K (ref. 41)), and notably in contrast to the other processes, its timescale depends on the excitation energy density and thus on the number of excited carriers19. The extracted time constants are 77±9 and 44±14 fs for 300 and 600 Jcm−3, respectively, and they are in fact in remarkably good agreement with the characteristic times needed for the buildup of screening, 68 and 54 fs, as estimated from the common formula for the plasma frequency19,46,47,48,49 (see Methods). The sub-100 fs sub-vibrational melting time, its distinct excitation density dependence and the accordance with free carrier screening then all point to a predominantly electronic phenomenon in 1T-TiSe2.
, which sets the timescale for the response of the correlated electron system to sudden changes44,45 (here W=80–400 meV corresponds to the width of the split-off Ta 5d band that straddles EF10,16,11). The melting of the Peierls gap, by contrast, takes at least one order of magnitude longer, about 226±38 fs. This value corresponds to half an oscillation cycle of the amplitude mode τamp/2=206 fs, a connection which is immediately visible in Fig. 3h, l and a clear indication for the structural origin of the gap. The same fingerprint of Peierls physics is seen in the melting of the energy gap in Rb:1T-TaS2, though on a shorter timescale (third row of Fig. 5). The melting time constant is 126±20 fs and agrees well with the structural timescale, τamp/2=117 fs. The suppression of the superstructure peak in 1T-TiSe2, on the other hand, is qualitatively different and cannot be classified as easily (second row of Fig. 5). The process is slower than electron hopping (τhop<1 fs for W≳ 1eV (ref. 20)), but faster than atomic motion (τamp/2>160 fs at 110 K (ref. 41)), and notably in contrast to the other processes, its timescale depends on the excitation energy density and thus on the number of excited carriers19. The extracted time constants are 77±9 and 44±14 fs for 300 and 600 Jcm−3, respectively, and they are in fact in remarkably good agreement with the characteristic times needed for the buildup of screening, 68 and 54 fs, as estimated from the common formula for the plasma frequency19,46,47,48,49 (see Methods). The sub-100 fs sub-vibrational melting time, its distinct excitation density dependence and the accordance with free carrier screening then all point to a predominantly electronic phenomenon in 1T-TiSe2.
Discussion
The systematic comparison of spectral responses under identical excitation conditions and the direct connection of the observed timescales to elementary processes involving the electrons and the lattice thus lead us to conclude that the puzzling CDW phases of Rb:1T-TaS2 and 1T-TiSe2 are dominated by electron–phonon interactions and by electron–electron interactions, respectively. In other words, Rb:1T-TaS2 is a Peierls insulator, whereas 1T-TiSe2 is indeed an excitonic insulator. To further support these claims, we will now briefly discuss our time-resolved ARPES results in the context of some important recent experimental findings on the same systems.
The key experimental advance of our study of the reference material 1T-TaS2 is to determine the electronic structure dynamics near the edge of the unreconstructed Brillouin zone, specifically in the region of momentum space in which the nesting-induced gap opens up to stabilize the periodic lattice distortion of the high-temperature incommensurate CDW phase (Fig. 2a,b). This is the region in which the primary structural component of the CDW in 1T-TaS2 can be probed, as confirmed by the vibrational timescale of the spectral response and consistent with the delayed response seen in paradigmatic CDW systems35,50. In a recent time-resolved ARPES study of 1T-TaS2 (ref. 18), by contrast, only the region around  was probed and, due to the better energy resolution compared with our experiment, a sub-vibrational collapse of the subband splitting at about 250 meV below EF (Fig. 2b) was observed. This region of momentum space therefore seems more appropriate to probe the secondary electronic component of the CDW in 1T-TaS2, which, however, may be intertwined with the Mott physics that emerges at the lock-in transition to the low-temperature commensurate CDW phase.
was probed and, due to the better energy resolution compared with our experiment, a sub-vibrational collapse of the subband splitting at about 250 meV below EF (Fig. 2b) was observed. This region of momentum space therefore seems more appropriate to probe the secondary electronic component of the CDW in 1T-TaS2, which, however, may be intertwined with the Mott physics that emerges at the lock-in transition to the low-temperature commensurate CDW phase.
The identification of Rb:1T-TaS2 as a Peierls insulator rests on the clear spectroscopic fingerprint of lattice dynamics that has been established for the reference 1T-TaS2. The time-resolved ARPES results fully corroborate the conclusions presented in a recent time-integrated ARPES study34. Because of the remarkably large and extended energy gap, the Rb:1T-TaS2 should in fact be classified as a Peierls insulator in the strong-coupling regime, in which Fermi surface nesting does not have a role anymore15,51.
The time-domain classification of 1T-TiSe2 as an excitonic insulator may be more controversial. The argument comprises not only the shortness of the order-parameter melting time but also its distinct fluence dependence19 and consistency with screening as the essential ingredient of the excitonic insulator instability1,52,53. Although we have previously advanced this argument19, the potential uniqueness of the spectral response has remained unclear. It is the context of time-resolved ARPES results obtained from CDW systems of the same family of compounds which now establishes this uniqueness and thus constitutes, in our view, the most conclusive evidence for the existence of the excitonic insulator phase in 1T-TiSe2 to date: the ultrafast spectral response of 1T-TiSe2 is clearly different from that of the paradigmatic Peierls insulator. This claim does not conflict with the complete phonon softening observed in recent inelastic X-ray scattering measurements54 because in an excitonic CDW instability the actual soft mode is a mixed exciton-phonon mode so that the electronically driven CDW is always accompanied by a periodic lattice distortion53,55. Regarding the energy density needed to melt the CDW nonthermally, however, our time-resolved ARPES results are inconsistent with recent time-resolved X-ray diffraction measurements41. This is an open issue, but we would argue that for an excitonic insulator, it is more appropriate to monitor the electronic order-parameter dynamics rather than the dynamics of the periodic lattice distortion which may in fact be regarded as an accidental byproduct of the excitonic insulator instability due to finite electron–phonon coupling55. Yet, we also emphasise that the melting dynamics we focus on here provides a handle only on the predominant character of the phase being destroyed, but not on the origin of the transition to this phase. Electron–phonon coupling may therefore still be an important trigger for CDW formation in 1T-TiSe2.
On a general level, our results then demonstrate that different types of long-range electronic order are indeed quenched on different characteristic timescales and that notably a CDW, being generally composed of a lattice charge density and of an electronic charge density52, can melt either on a structural timescale, as in the Peierls insulator Rb:1T-TaS2, or on an electronic timescale, as in the excitonic insulator 1T-TiSe2, or on both timescales, as in the Peierls-Mott insulator 1T-TaS2 (ref. 18), depending on which density component or underlying interaction is the dominant one (at the probed momentum). The observed hierarchy of melting times ultimately seems to reflect three different non-thermal destruction mechanisms following photoexcitation (Fig. 1d): the Mott insulator collapses due to an ultrafast non-equilibrium rearrangement of the electronic states taking place on the elementary timescale of electron hopping44, the excitonic insulator breaks down because the Coulomb attraction causing electrons and holes to form excitons is screened by the added free carriers as soon as screening has built up on the timescale of the inverse plasma frequency19, and the Peierls insulator melts via a displacive excitation of coherent atomic motions resulting in a suppressed periodic lattice distortion within about half an oscillation of the amplitude mode40.
In conclusion, the direct, momentum-selective and systematic observation of electronic order parameter dynamics on the relevant timescales, as demonstrated here using time-resolved ARPES, offers a powerful and intuitive approach to assess the relative contributions of electron–electron and electron–phonon interactions to electronic order in momentum space: the very timescale on which the dynamics takes place, together with its excitation density dependence, uncovers the dominant interaction. The approach may help not only to capture the intermediate grades between pure Mott, Peierls and excitonic insulators but also to distinguish different types of gaps in, for example, unconventional superconductors.
Methods
Sample preparation
1T-TaS2 and 1T-TiSe2 single crystals were grown from high purity elements by chemical vapour transport using iodine as transport agent. Sample quality was checked by electronic transport measurements. In the photoemission experiments, all samples were cleaved under ultrahigh vacuum conditions at room temperature. The Rb:1T-TaS2 samples were prepared in situ by depositing Rb for 300 s on freshly cleaved 1T-TaS2 surfaces at room temperature (using a SAES getters dispenser operating at a current of 6 A and a working distance of about 3 cm).
Photoemission measurements
Time-integrated ARPES measurements were performed at beamline 7.0.1 of the Advanced Light Source at Berkeley employing a Scienta R4000 spectrometer. Typical effective energy and angle resolutions were 90 meV and 0.2°, respectively. The femtosecond time-resolved ARPES experiments were done at the University of Kiel using a SPECS Phoibos 150 analyser. Here, the total energy resolution was 400 meV, mainly governed by the spectral broadening of the femtosecond XUV pulses, and the angle resolution was estimated to be 1.5°. Before each measurement run, the sample orientation and electronic gap sizes were checked by time-integrated ARPES using a He discharge lamp (hv=21.22 eV). The equilibrium sample temperature was 110 K for all reported time-resolved ARPES data.
The basic light source used for pump-probe photoemission was a 3-kHz Ti:sapphire amplifier system (KMLabs Dragon pumped by a Spectra Physics Empower 30) delivering near-infrared pulses at 790 nm wavelength, 1.2 mJ pulse energy and 30 fs (FWHM) pulse duration (representing the major contribution to the effective time resolution of the experiment). For photoemission, 90% of the pulse energy was used to generate high harmonic femtosecond XUV pulses in an argon-filled hollow-fibre waveguide (KMLabs XUUS). A pair of multilayer mirrors (with a total reflectivity of 13%) selected the 27th harmonic (hv=43 eV) out of the harmonic spectrum and focused it onto the sample mounted in the ultrahigh vacuum system. From infrared-XUV cross-correlation measurements, the pulse duration of the XUV probe pulses was estimated to be shorter than 10 fs. The remaining 10% of the amplifier output were available for photoexcitation of the samples. The s-polarized pump pulses impinged on the sample under 45°, except for measurements around the point of 1T-TaS2 for which the incidence angle was about 25° relative to the surface normal. For pristine 1T-TaS2 and 1T-TiSe2, the absorbed energy densities and corresponding temperature increases in the excited sample volumes were determined from the measured incident pump fluences using reflection coefficients of 0.41 (ref. 56) and 0.65 (ref. 57), penetration depths of 36 (ref. 56) and 15 nm (ref. 57), specific heat β values of 12.6 (ref. 58) and 9.45 Jm−3 K−4 (ref. 59), and Debye temperatures of 237 (ref. 58) and 251 K (ref. 59), respectively. In the case of Rb:1T-TaS2, we assumed that the system can be reasonably described by the optical and specific heat parameters of pure 1T-TaS2.
Data analysis
The temporal evolutions of the ARPES intensities in the energy windows of interest were fitted with a model function composed of the Gaussian error function to capture the fast initial collapse and a single exponential to model the slower relaxation dynamics. The finite time resolution of the experiment was modelled by a convolution with a Gaussian of 32 fs FWHM. The stated melting times correspond to the FWHM of the derivative of the Gaussian error function.
The fluence dependent screening times in 1T-TiSe2 were estimated from the common formula for the plasma frequency,

using a relative electric constant of εγ=19 (ref. 60), an effective isotropic electron mass of  (ref. 13), a free carrier density of n0=0.1 × 1019 cm−3 (ref. 60), an assumed quantum efficiency (QE) of 0.1 and a photon density n derived from the absorbed energy density (ε0: electric constant; e0: elementary charge; me: electron mass).
(ref. 13), a free carrier density of n0=0.1 × 1019 cm−3 (ref. 60), an assumed quantum efficiency (QE) of 0.1 and a photon density n derived from the absorbed energy density (ε0: electric constant; e0: elementary charge; me: electron mass).
Additional information
How to cite this article: Hellmann, S. et al. Time-domain classification of charge-density-wave insulators. Nat. Comm. 3:1069 doi: 10.1038/ncomms2078 (2012).
References
Mott, N. F. The transition to the metallic state. Phil. Mag. 6, 287–309 (1961).
Van Zandt, L. L. & Honig, J. M. Theories pertaining to the semiconductor-metal transition in crystals. Annu. Rev. Mater. Sci. 4, 191–220 (1974).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039–1263 (1998).
Edwards, P. P., Johnston, R. L., Rao, C. N. R., Tunstall, D. P. & Hensel, F. The metal-insulator-transition: a perspective. Phil. Trans. R. Soc. Lond. A 356, 5–22 (1998).
Gebhard, F. The Mott Metal-Insulator Transition (Springer, Berlin, 2000).
Wilson, J. A., Di Salvo, F. J. & Mahajan, S. Charge-density waves and superlattices in the metallic layered transition metal dichalcogenides. Adv. Phys. 24, 117–201 (1975).
Friend, R. H. & Yoffe, A. D. Electronic properties of intercalation complexes of the transition metal dichalcogenides. Adv. Phys. 36, 1–94 (1987).
Dardel, B. et al. Temperature-dependent pseudogap and electron localization in 1T-TaS2 . Phys. Rev. B 45, 1462–1465 (1992).
Perfetti, L., Gloor, T. A., Mila, F., Berger, H. & Grioni, M. Unexpected periodicity in the quasi-two-dimensional Mott insulator 1T-TaS2 revealed by angle-resolved photoemission. Phys. Rev. B 71, 153101 (2005).
Rossnagel, K. & Smith, N. V. Spin-orbit coupling in the band structure of reconstructed 1T-TaS2 . Phys. Rev. B 73, 073106 (2006).
Freericks, J. K., Krishnamurthy, H. R., Ge, Y., Liu, A. Y. & Pruschke, T. Theoretical description of time-resolved pump/probe photoemission in TaS2: a single-band DFT+DMFT(NRG) study within the quasiequilibrium approximation. Phys. Status Solidi B 246, 948–954 (2009).
Rossnagel, K., Kipp, L. & Skibowski, M. Charge-density-wave phase transition in 1T-TiSe2: excitonic insulator versus band-type Jahn-Teller mechanism. Phys. Rev. B 65, 235101 (2002).
Kidd, T. E., Miller, T., Chou, M. Y. & Chiang, T.- C Electron-hole coupling and the charge density wave transition in TiSe2 . Phys. Rev. Lett. 88, 226402 (2002).
Cercellier, H. et al. Evidence for an excitonic insulator phase in 1T-TiSe2 . Phys. Rev. Lett. 99, 146403 (2007).
Rossnagel, K. On the origin of charge-density waves in select layered transition-metal dichalcogenides. J. Phys. Condens. Matter 23, 213001 (2011).
Perfetti, L. et al. Time evolution of the electronic structure of 1T-TaS2 through the insulator-metal transition. Phys. Rev. Lett. 97, 067402 (2006).
Perfetti, L. et al. Femtosecond dynamics of electronic states in the Mott insulator 1T-TaS2 by time resolved photoelectron spectroscopy. New J. Phys. 10, 053019 (2008).
Petersen, J. C. et al. Clocking the melting transition of charge and lattice order in 1T-TaS2 with ultrafast extreme-ultraviolet angle-resolved photoemission spectroscopy. Phys. Rev. Lett. 107, 177402 (2011).
Rohwer, T. et al. Collapse of long-range charge order tracked by time-resolved photoemission at high momenta. Nature 471, 490–493 (2011).
Monney, C., Battaglia, C., Cercellier, H., Aebi, P. & Beck, H. Exciton condensation driving the periodic lattice distortion of 1T-TiSe2 . Phys. Rev. Lett. 106, 106404 (2011).
Morosan, E. et al. Superconductivity in CuxTiSe2 . Nat. Phys. 2, 544–550 (2006).
Sipos, B. et al. From Mott state to superconductivity in 1T-TaS2 . Nat. Mater. 7, 960–965 (2008).
Kusmartseva, A. F., Sipos, B., Berger, H., Forró, L. & Tutiš, E. Pressure induced superconductivity in pristine 1T-TiSe2 . Phys. Rev. Lett. 103, 236401 (2009).
Fazekas, P. & Tosatti, E. Electrical, structural and magnetic properties of pure and doped 1T-TaS2 . Philos. Mag. B 39, 229–244 (1979).
Di Salvo, F. J., Moncton, D. E. & Waszczak, J. V. Electronic properties and superlattice formation in the semimetal TiSe2 . Phys. Rev. B 14, 4321–4328 (1976).
Yoshida, Y. & Motizuki, K. Electron lattice interaction and lattice instability of 1T-TiSe2 . J. Phys. Soc. Jpn 49, 898–905 (1980).
Hughes, H. P. Structural distortion in TiSe2 and related materials—a possible Jahn-Teller effect? J. Phys. C Solid State Phys. 10, L319 (1977).
Whangbo, M.- H. & Canadell, E. Analogies between the concepts of molecular chemistry and solid-state physics concerning structural instabilities. Electronic origin of the structural modulations in layered transition metal dichalcogenides. J. Am. Chem. Soc. 114, 9587–9600 (1992).
White, R. M. & Lucovsky, G. Suppression of antiferroelectricity in TiSe2 by excess carriers. Nuovo Cimento B 38, 280–289 (1977).
Wilson, J. A. Modelling the contrasting semimetallic characters of TiS2 and TiSe2 . Phys. Status Solidi B 86, 11–36 (1978).
van Wezel, J., Nahai-Williamson, P. & Saxena, S. S. Exciton-phonon-driven charge density wave in TiSe2 . Phys. Rev. B 81, 165109 (2010).
Clark, W. B. & Williams, P. M. Charge density waves in intercalated 1T-TaS2 . Philos. Mag. 35, 883–899 (1977).
Rossnagel, K., Rotenberg, E., Koh, H., Smith, N. V. & Kipp, L. Continuous tuning of electronic correlations by alkali adsorption on layered 1T-TaS2 . Phys. Rev. Lett. 95, 126403 (2005).
Rossnagel, K. Suppression and emergence of charge-density waves at the surfaces of layered 1T-TiSe2 and 1T-TaS2 by in situ Rb deposition. New J. Phys. 12, 125018 (2010).
Schmitt, F. et al. Transient electronic structure and melting of a charge density wave in TbTe3 . Science 321, 1649–1652 (2008).
Schmitt, F. et al. Ultrafast electron dynamics in the charge density wave material TbTe3 . New J. Phys. 13, 063022 (2011).
Rundquist, A. et al. Phase matching of soft-X-ray harmonic emission in hollow-core fibers. Science 280, 1412–1415 (1998).
Mathias, S., Miaja-Avila, L., Murnane, M. M., Kapteyn, H., Aeschlimann, M. & Bauer, M. Angle-resolved photoemission spectroscopy with a femtosecond high harmonic light source using a two-dimensional imaging electron analyzer. Rev. Sci. Instrum. 78, 083105 (2007).
Hellmann, S. et al. Ultrafast melting of a charge-density wave in the Mott insulator 1T-TaS2 . Phys. Rev. Lett. 105, 187401 (2010).
Eichberger, M. et al. Snapshots of cooperative atomic motions in the optical suppression of charge density waves. Nature 468, 799–802 (2010).
Möhr-Vorobeva, E. et al. Nonthermal melting of a charge density wave in TiSe2 . Phys. Rev. Lett. 107, 036403 (2011).
Kusar, P. et al. Anharmonic order-parameter oscillations and lattice coupling in strongly driven 1T-TaS2 and TbTe3 charge-density-wave compounds: a multiple-pulse femtosecond laser spectroscopy study. Phys. Rev. B 83, 035104 (2011).
Demsar, J., Forró, L., Berger, H. & Mihailovic, D. Femtosecond snapshots of gap-forming charge-density-wave correlations in quasi-two-dimensional dichalcogenides 1T-TaS2 and 2H-TaSe2 . Phys. Rev. B 66, 041101(R) (2002).
Moritz, B., Devereaux, T. P. & Freericks, J. K. Time-resolved photoemission of correlated electrons driven out of equilibrium. Phys. Rev. B 81, 165112 (2010).
Wall, S. et al. Quantum interference between charge excitation paths in a solid-state Mott insulator. Nat. Phys. 7, 114–118 (2010).
El Sayed, K., Schuster, S., Haug, H., Herzel, F. & Henneberger, K. Subpicosecond plasmon response: buildup of screening. Phys. Rev. B 49, 7337–7344 (1994).
Bányai, L., Vu, Q. T., Mieck, B. & Haug, H. Ultrafast quantum kinetics of time-dependent rpa-screened coulomb scattering. Phys. Rev. Lett. 81, 882–885 (1998).
Huber, R. et al. How many-particle interactions develop after ultrafast excitation of an electron-hole plasma. Nature 414, 286–289 (2001).
Huber, R. et al. Femtosecond formation of coupled phonon-plasmon modes in inp: ultrabroadband THz experiment and quantum kinetic theory. Phys. Rev. Lett. 94, 027401 (2005).
Tomeljak, A. et al. Dynamics of photoinduced charge-density-wave to metal phase transition in K0.3MoO3 . Phys. Rev. Lett. 102, 066404 (2009).
Aruga, T. Surface Peierls transition on Cu(001) covered with heavier p-block metals. Surf. Sci. Rep. 61, 283–302 (2006).
Halperin, B. I. & Rice, T. M. The excitonic state at the semiconductor-semimetal transition. Solid State Phys. 21, 115–192 (1968).
Kohn, W. Metals and Insulators in Many Body Physics (eds DeWitt, C. and Balian R.) 353–411 (Gordon and Breach, New York, 1968).
Weber, F. et al. Electron-phonon coupling and the soft phonon mode in TiSe2 . Phys. Rev. Lett. 107, 266401 (2011).
Tosatti, E. Electronic Superstructures of Semiconductor Surfaces and of Layered Transition-Metal Compounds in Festkörperprobleme (Advances in Solid State Physics) Vol. 15: (ed Queisser, H.J.) 113–147 (Vieweg, Braunschweig, 1975).
Beal, A. R., Hughes, H. P. & Liang, W. Y. The reflectivity spectra of some group VA transition metal dichalcogenides. J. Phys. C Solid State Phys. 8, 4236–4248 (1975).
Bayliss, S. C. & Liang, W. Y. Reflectivity, joint density of states and band structure of group IVb transition-metal dichalcogenides. J. Phys. C Solid State Phys. 18, 3327–3335 (1985).
Schwall, R. E., Stewart, G. R. & Geballe, T. H. Low-temperature specific heat of layered compounds. J. Low Temp. Phys. 22, 557–567 (1976).
Craven, R. A., Di Salvo, F. J. & Hsu, F. S. L. Mechanisms for the 200 K transition in TiSe2: a measurement of the specific heat. Solid State Commun. 25, 39–42 (1978).
Li, G. et al. Semimetal-to-semimetal charge density wave transition in 1T-TiSe2 . Phys. Rev. Lett. 99, 027404 (2007).
Acknowledgements
This work was primarily supported by the German Federal Ministry of Education and Research (Project Nos. 05K10FK1 and 05K10FK4). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy, under Contract No. DE-AC02-05CH11231. A.C., M.M. and H.K. acknowledge support from the NSF Physics Frontier Centers and used facilities provided by NSF EUV ERC.
Author information
Authors and Affiliations
Contributions
Design of the study was done by S.H., T.R., M.B. and K.R. Operation of the laser system was carried out by T.R. and A.S. Time-resolved ARPES measurements were done by S.H., T.R., K.H., C.S. and A.S. Conventional ARPES measurements were done by M.K. and K.R. Mirror design, fabrication and characterization were done by A.C. Technical support and conceptual advice were given by M.M., H.K. and L.K. Data analysis was done by S.H., M.K. and K.R. Manuscript preparation was done by S.H. and K.R., with input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Hellmann, S., Rohwer, T., Kalläne, M. et al. Time-domain classification of charge-density-wave insulators. Nat Commun 3, 1069 (2012). https://doi.org/10.1038/ncomms2078
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms2078
This article is cited by
-
Endotaxial stabilization of 2D charge density waves with long-range order
Nature Communications (2024)
-
Time, momentum, and energy resolved pump-probe tunneling spectroscopy of two-dimensional electron systems
Nature Communications (2023)
-
Crystallization of polarons through charge and spin ordering transitions in 1T-TaS2
Nature Communications (2023)
-
In-gap band formation in a periodically driven charge density wave insulator
Communications Physics (2023)
-
Strong correlations in two-dimensional transition metal dichalcogenides
Science China Physics, Mechanics & Astronomy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
