
Abstract
Water ice dissociates into a superionic solid at high temperature (>2,000 K) and pressure, where oxygen forms the lattice, but hydrogen diffuses completely. At low temperature, however, the dissociation into an ionic ice of hydronium (H3O)+ hydroxide (OH)− is not expected because of the extremely high energy cost (∼1.5 eV) of proton transfer between H2O molecules. Here we show the pressure-induced formation of a partially ionic phase (monoclinic P21 structure) consisting of coupled alternate layers of (OH)δ− and (H3O)δ+ (δ=0.62) in water ice predicted by particle-swarm optimization structural search at zero temperature and pressures of >14 Mbar. The occurrence of this ionic phase follows the break-up of the typical O–H covalently bonded tetrahedrons in the hydrogen symmetric atomic phases and is originated from the volume reduction favourable for a denser structure packing.
Similar content being viewed by others


Introduction
Water ice belongs to an important group of archetypal binary compounds with a large abundance in the interiors of giant planets1,2. The cores of Uranus, Neptune, Saturn and Jupiter are assumed to contain largely of water ice at pressures up to 40 Mbar (ref. 1). Thus, the ultra-high pressure studies on the structure properties of ice is of crucial importance for the understanding of the physics and chemistry of planetary interiors and even origin of their magnetic field distribution3,4,5. However, the conditions as occurring in the cores of giant plants are not directly experimentally accessible, as an alternative ab initio simulations are able to explore states of matters under such extreme conditions.
Ice has a very rich phase diagram. Till now, at least 15 solid phases have been identified experimentally6,7 and 4 other high-pressure phases were theoretically predicted8,9. Experimentally, all crystalline phases except ice X are found to consist of water molecules (each has two O–H covalent bonds) connected by hydrogen bonds. At low temperature and pressures above 70 GPa, ice dissociates and stabilizes at phase X, an atomic crystal having the symmetrical hydrogen bonds with each O atom covalently bonded to four H atoms. No higher-pressure phases beyond ice X have been experimentally verified up to the highest pressure 2.1 Mbar investigated so far. Theoretically, first-principles simulations established that an orthorhombic Pbcm phase surpasses ice X at ∼3 Mbar (ref. 8) and is energetically more stable than the earlier anti-fluorite structure10. Very recently, three 0 K crystalline Pbca, Pnma and Cmcm structures stable above 7.6 Mbar are proposed by a simulated annealing method9. These later theoretical atomic structures of post-ice X share the similarity with ice X, possessing fourfold O–H covalent bonds.
Of particular interest is the recent finding of superionic ice at high temperatures (>2,000 K) and high pressures5,6, where oxygen remains fixed at the lattice, but hydrogen atoms diffuse completely. This superionicity is a clear temperature-driven phenomenon, allowing the break-up of the O–H covalent bond. At low temperature, however, the hydrogen atoms remain covalently bonded with oxygen where hydrogen transfer between two H2O molecules is energetically very unfavourable with a large energy cost (for example, 1.5 eV in solid11). Therefore, the finding of ionic forms in ice at low temperature is scarce. To date, the dissociation of a H2O molecule in liquid water into (OH)− and (H3O)+ at ambient pressure, and its high pressure enhancement have been reported12,13,14, but it is, in general, rare, short-lived and dynamically controlled.
Here we report the direct prediction of formation of a partially ionic phase in water ice consisting of coupled alternate layers of (OH)δ− and (H3O)δ+ (δ=0.62) through our recently developed particle swarm optimization (PSO) algorithm for crystal structure prediction15 at zero temperature and pressures (>14 Mbar). We also predict that a tetrahedrally O–H bonded covalent ice (tetragonal I-42d structure) containing highly distorted O–H tetrahedrons surpasses other known ice phases above 8.1 Mbar, bridging the hydrogen symmetric atomic phases with the ionic phase.
Results
Crystal structures
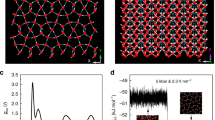
Our structure searches through CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) code15 with system sizes up to eight H2O formula units (f.u.) per simulation cell through both global and local PSO schemes, were performed at 2, 5, 7, 10, 15 and 20 Mbar. The experimental ice X and earlier theoretical Pbcm, Pbca, Pnma and Cmcm structures were all successfully reproduced in certain pressure ranges through both PSO techniques, validating our methodology in application to dense ice. We newly predicted two energetically favourable structures: a tetragonal I-42d (16 f.u. or cell, Fig. 1a; Supplementary Table S1) and a monoclinic P21 (4 f.u. per cell, Fig. 1b; Supplementary Table S1). I-42d structure containing two interpenetrating hydrogen-bonded networks, which can only be found by the more efficient local PSO scheme, shares the bonding similarity to earlier Pbcm and Pbca structures with each O atom bonded with four H atoms, but achieves denser structure packing with stronger bond–angle distortions in the tetrahedrons (for example, at 10 Mbar the H–O–H bond–angle distribution in I-42d structure extends from 92 to 127° instead of from 86 to 127° in Pbca). Intriguingly, in P21 structure, the structural feature of O–H tetrahedrons is no longer preserved. Instead, we find the surprising occurrence of alternate layers of OH and H3O units (Fig. 1b), implying the ionic nature of this solid. It should be noted that after the submission of this work, we became aware of an unpublished paper (arXiv:1106.1941v1) by McMahon, reporting the similar P21 structure and a different lower pressure structure Pmc21, which, however, is energetically less favourable than our I-42d structure.

(a) Crystal structure of I-42d viewed along the c axis. (b) Crystal structure of P21 viewed along a axis. The large spheres represent the oxygen atoms whereas the small ones are for hydrogen atoms. The atoms and O–H distances are labelled in accordance with the notation in Table 1. (c) Isosurface of ELF for I-42d structures at 14 Mbar with the ELF value of 0.75. (d) Isosurface of ELF for P21 structures at 14 Mbar with the ELF value of 0.75.
Structural stability
The enthalpies of the predicted stable phases, calculated at higher levels of accuracy, are plotted as a function of pressure in Figure 2a. It is seen that ice transforms from Pbcm to Pbca at 7.6 Mbar (Fig. 2b), in excellent agreement with earlier theoretical calculations9. The Pbca structure is stable at 7.6–8.1 Mbar, above which the I-42d structure takes over and exists in a large pressure range till 16 Mbar, where P21 structure becomes most energetically favourable up to 20 Mbar, the highest pressure studied. Note that the zero-point energy (ZPE) is not included in the enthalpy calculations, which can be very large owing to the small atomic masses of H and O atoms. Indeed, our estimated ZPEs for Pbca and I-42d structures at 10 Mbar by a quasi-harmonic model16 have large values of 1.105 eV f.u.−1 and 1.110 eV f.u.−1, respectively. However, their difference is very small, giving a negligible modification on the transition pressure of Pbca→I-42d. Interestingly, P21 structure has a much smaller ZPE (1.148 eV f.u.−1) than that of I-42d structure (1.229 eV f.u.−1) at 14 Mbar, lowering the I-42d→P21 transition pressure from 16 to 14 Mbar (Fig. 2c.) The physical origin of smaller ZPE in P21 structure is largely related to the gross weaker O–H covalent bonding as evidenced by the averaged larger O–H bond lengths (for example, at 14 Mbar, the average O–H bond length is 1.03 Å for P21 structure to compare with 0.97 Å for I-42d structure). It should be noted that both I-42d and P21 structures are dynamically stable in their stability fields (Supplementary Fig. S1).

(a) Calculated enthalpies (H) per H2O unit as a function of pressure (P) for various structures with respect to Pbcm structure. The green, blue and black lines indicate the enthalpies for Pbca, Pnma and Cmcm, respectively. The enthalpies of I-42d and P21 structures are marked by the red and pink lines, respectively. (b) The enthalpies (7.2–9 Mbar) of Pbca and I-42d structures with respect to Pbcm in more details. (c) The enthalpies of P21 structure with respect to I-42d structure with ZPE included.
Chemical bondings
The unexpected formation of OH and H3O units invites us to perform a thorough analysis on the chemical bondings of P21 structure. We first calculated the electron localization functions (ELF), known to be an informative tool to distinguish different bonding interactions in solid17. The isosurface plots at ELF=0.75 (a typical good number for characterization of covalent bondings)17 clearly illustrate the covalent bonding nature of the I-42d structure (Fig. 1c) and confirm the formation of OH and H3O units in P21 structure (Fig. 1d; Supplementary Fig. S2). However, we also noticed a small charge distribution localized in between OH and H3O, an indicative of their covalent interaction. We subsequently performed a topological analysis of the static electron density through Bader's quantum theory of atoms-in-molecules18, which has been successfully applied to the determination of bonding interactions through the values of the density and its Laplacian at bond critical points. The calculated data are summarized in Table 1. Again, the analysis gives strong evidence on the formation of OH and H3O units as indicted by the very negative values of the Laplacian of charge density at the critical points. In agreement with above ELF results, our calculation also supports the covalent interaction (though relatively weak) between OH and H3O layers as seen from the noticeable negative Laplacian value (Table 1) between O1 and H2/H3/H4 (d2, d3 and d4 in Fig. 1b). This interaction is understandable because P21 structure is a highly packed structure at such an extreme pressure of 14 Mbar (at higher pressure 20 Mbar, this interaction becomes even stronger as shown in Table 1). The two identities are too close to not be covalently interacted. However, this covalent coupling is, in fact, not intrinsic as we find the interaction decreases significantly with lowering pressure to 7 Mbar (Table 1), whereas the extraordinarily strong O–H bondings within OH and H3O units remain nearly unaltered, and at an extreme case by extrapolation to about 2 Mbar, no interaction was found at all. To support our argument, we have theoretically compressed the perfect ionic NH4+NH2− solid derived from NH3 (ref. 19) and found a similar covalent interaction of NH4+ and NH2− at a pressure 7 Mbar. On the basis of Bader theory18, the approximate charge values of oxygen and hydrogen ions of P21 structure are calculated at 14 Mbar as listed in Table 2. We found different charges for inequivalent oxygen and hydrogen ions as determined by their distinct chemical environments. It is seen that the charge transfer from H3O to OH is about 0.62e at 14 Mbar, illustrating the ionic nature of P21 structure with a notation of (OH)δ− (H3O)δ+ (δ=0.62).
Electronic structures
The calculated electronic band structures (Supplementary Fig. S3) for I-42d and P21 structures reveal that the two phases are both insulators with large band gaps (>5.0 eV at 14 Mbar for P21 structure), which show weak dependence on pressure (for example, at a rate of −0.135 eV Mbar−1 for P21 structure). Considering also that density-functional calculations usually underestimate the band gaps, we expect that the low-temperature metallization of ice is only possible at pressures much higher than 20 Mbar. Our results are in apparently contrast to the earlier proposal on the metallicity of ice at 15.5 Mbar (ref. 9).
Discussion
Above 3 Mbar, ice X transforms into Pbcm, Pbca, and I-42d structures, which all contain distorted O–H tetrahedrons (oxygen is in the centre, whereas hydrogen atoms occupy the tetrahedral sites) where hydrogen atoms are squeezed out of midpoint between oxygen atoms. These transformations follow the continuous distortion of O–H tetrahedrons with severer H deviations (for example, the enlarged H–O–H angles), but the tetrahedrons remain. Eventually, at the formation of P21 structure, the typical O–H tetrahedrons in the atomic phases collapse completely (Fig. 1a,b). We contribute the phase transformations to the needs for denser structure packing driven by pressure (Supplementary Fig. S4). Our calculations show that the sphere packing efficiency at 14 Mbar increases continuously in the order of 25.5%→29.4%→32.1%→33.93%→35.44% for ice X, Pbcm, Pbca, I-42d, and P21 structure, respectively. As is known that Gibbs free energy reduces to H=U+PV at 0 K, where U, P and V are the static energy, pressure and volume per formula unit, respectively. The high pressure competition between U and PV dominate the phase transitions. Although, the first order transitions of Pbca→I-42d→P21 have rather small 1.8% and 1.9% volume drops, respectively, these volume reductions are critical to initiate the phase transitions. It is calculated that although P21 structure is 0.59 eV per H2O unit higher in U than I-42d structure at 17 Mbar, its PV term is 0.62 eV lower owing to the volume reduction associated with a higher packing efficiency.
The pressure-induced formation of ionic solid on molecular systems has been predicted theoretically for NH3 (ref. 19), H2S20 and NH3·H2O21 and observed experimentally for N2O22. The dissociation of H2S into SH− and SH3+ is short lived and dynamically controlled20, similar to that in liquid water12,13,14,23. The stabilization of NO3−NO+ solid compressed from N2O violates the conservation of chemical composition and there temperature has had a significant role on its formation22. Our finding in some ways resembles with the ionic mechanism of NH3 (ref. 19) and NH3·H2O21 solids. What makes the ionicity in H2O unique is the existence of covalent coupling between OH and H3O, leading to a partial ionicity. Notably, the ionic pressure (∼14 Mbar) in H2O is considerably higher than ∼1 Mbar in NH3 (ref. 19) and 0.05 Mbar in NH3·H2O21. The physical origin behind this high ionic pressure is stemmed from the much larger energy barrier (∼1.5 eV11) of proton transfer between two H2O molecules.
The current findings of high-pressure phases of I-42d and P21 structures have enriched the chemical bondings of ice, and substantially extended the low-temperature phase diagram of ice to the pressure ranges of 8.1–20 Mbar, relevant to the interiors of giant planets. Our results may be useful as the initial low-temperature structures to build up the constituent model for planetary interiors at high temperature. Our prediction of partially ionic ice is unusual in view of the existence of large energy barriers of hydrogen transfer between two H2O molecules, but highlights the critical role pressure had in overcoming the energy barriers. Energetically, the tuning effect of PV term with respect to static energy U is substantial in light of volume reduction preferable for denser structural packing. In the same manner, ionicity might be expected in other molecular systems (for example, CO2, NO2 and so on), not at low but extreme pressures.
Methods
Crystal structure prediction
Our structure search simulations are performed through the CALYPSO method24 as implemented in CALYPSO code15, which is specially designed for global structural minimization unbiased by any known structural information. Our approach is based on a global minimization of free-energy surfaces merging ab initio total-energy calculations via PSO technique24. Our CALYPSO method has been benchmarked on various known systems24 with various chemical bondings and had several successful prediction of high-pressure structures of Li, Mg and Bi2Te3 (refs 25,26,27), among which the insulating Aba2–40 (or oC40) structure of Li and the two low-pressure monoclinic structures of Bi2Te3 have been confirmed by independent experiments27,28. We have adopted a local PSO scheme, which was designed to avoid the premature structure convergence by maintaining multiple attractors and to allow the exploration of larger space of free-energy surfaces. Therefore, the local PSO is computationally more demanding, but more effective than global PSO technique, particularly when simulation sizes are larger than 24–30 atoms per cell where the free-energy landscape is more complex. Detailed description of the method and predictions can be found in the Supplementary Methods.
Total energy calculations
Total energy calculations were performed in the framework of density functional theory within the Perdew–Burke–Ernzerhof29 parameterization of generalized gradient approximation30 as implemented in the VASP (Vienna Ab Initio simulation package) code31. The projector-augmented wave (PAW) method32,33 was adopted with the PAW potentials taken from the VASP library where 1s1 and 2s22p4 are treated as valence electrons for H and O atoms, respectively. The validity of the used PAW potential at currently studied pressures (<20 Mbar) has earlier been carefully checked by comparing with the full-potential all-electron calculations through Exciting code9. The use of the plane-wave kinetic energy cutoff of 1,200 eV and dense k-point sampling34, adopted here, were shown to give excellent convergence of total energies.
Additional information
How to cite this article: Wang, Y. et al. High pressure partially ionic phase of water ice. Nat. Commun. 2:563 doi: 10.1038/ncomms1566 (2011).
References
Guillot, T. Interiors of giant planets inside and outside the solar system. Science 286, 72–77 (1999).
Lissauer, J. J. Extrasolar planets. Nature 419, 355–358 (2002).
French, M., Mattsson, T. R., Nettelmann, N. & Redmer, R. Equation of state and phase diagram of water at ultrahigh pressures as in planetary interiors. Phys. Rev. B 79, 054107 (2009).
French, M., Mattsson, T. R. & Redmer, R. Diffusion and electrical conductivity in water at ultrahigh pressures. Phys. Rev. B 82, 174108 (2010).
Cavazzoni, C., Chiarotti, G. L., Scandolo, S., Tosatti, E., Bernasconi, M. & Parrinello, M. Superionic and metallic states of water and ammonia at giant planet conditions. Science 283, 44–46 (1999).
Petrenko, V. & Whitworth, R. Physics of Ice, Oxford University Press, USA, 1999.
Salzmann, C. G., Radaelli, P. G., Mayer, E. & Finney, J. L. Ice XV: a new thermodynamically stable phase of ice. Phys. Rev. Lett. 103, 105701 (2009).
Benoit, M., Bernasconi, M., Focher, P. & Parrinello, M. New high-pressure phase of ice. Phys. Rev. Lett. 76, 2934–2936 (1996).
Militzer, B. & Wilson, H. F. New phases of water ice predicted at megabar pressures. Phys. Rev. Lett. 105, 195701 (2010).
Demontis, P., LeSar, R. & Klein, M. New high-pressure phases of ice. Phys. Rev. Lett. 60, 2284–2287 (1988).
Liebman, J. F. Existence and estimated enthalpies of formation of ammonium hydroxide, hydronium amide, and some related species. Struct. Chem. 8, 313–315 (1997).
Holmes, N., Nellis, W., Graham, W. & Walrafen, G. Spontaneous Raman scattering from shocked water. Phys. Rev. Lett. 55, 2433–2436 (1985).
Schwegler, E., Galli, G., Gygi, F. & Hood, R. Q. Dissociation of water under pressure. Phys. Rev. Lett. 87, 265501 (2001).
Goncharov, A. F. et al. Dynamic ionization of water under extreme conditions. Phys. Rev. Lett. 94, 125508 (2005).
Ma, Y., Wang, Y., Lv, J. & Zhu, L. CALYPSO code, http://nlshm-lab.jlu.edu.cn/~calypso.html.
Ma, Y. & Tse, J. S. Ab initio determination of crystal lattice constants and thermal expansion for germanium isotopes. Solid State Commun. 143, 161–165 (2007).
Savin, A., Nesper, R., Wengert, S. & Fasler, T. F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 36, 1808–1832 (1997).
Bader, R. F. W. Atoms in Molecules: A Quantum Theory Vol. 22, Clarendon Press, Oxford, UK, (1990).
Pickard, C. J. & Needs, R. Highly compressed ammonia forms an ionic crystal. Nature Mater. 7, 775–779 (2008).
Rousseau, R., Boero, M., Bernasconi, M., Parrinello, M. & Terakura, K. Ab initio simulation of phase transitions and dissociation of H2S at high pressure. Phys. Rev. Lett. 85, 1254–1257 (2000).
Fortes, A., Brodholt, J., Wood, I., Vo adlo, L. & Jenkins, H. Ab initio simulation of ammonia monohydrate (NH·HO) and ammonium hydroxide (NH4OH). J. Chem. Phys. 115, 7006 (2001).
Somayazulu, M. et al. Novel broken symmetry phase from N2O at high pressures and high temperatures. Phys. Rev. Lett. 87, 135504 (2001).
Giguere, P. A. The great fallacy of the H+ ion: and the true nature of H3O+. J. Chem. Educ. 56, 571 (1979).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Lv, J., Wang, Y., Zhu, L. & Ma, Y. Predicted novel high-pressure phases of lithium. Phys. Rev. Lett. 106, 015503 (2011).
Li, P., Gao, G., Wang, Y. & Ma, Y. Crystal structures and exotic behavior of magnesium under pressure. J. Phys. Chem. C 114, 21745–21749 (2010).
Zhu, L., Wang, H., Wang, Y., Lv, J., Ma, Y., Cui, Q. & Zou, G. Substitutional alloy of Bi and Te at high pressure. Phys. Rev. Lett. 106, 145501 (2011).
Guillaume, C. L. et al. Cold melting and solid structures of dense lithium. Nature Phys. 7, 211–214 (2011).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244 (1992).
Kresse, G. & Furthmuler, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Acknowledgements
We acknowledge the funding supports from the National Natural Science Foundation of China under grant Nos. 11025418 and 91022029, and the China 973 Program under Grant No. 2011CB808200. Part of the calculations was performed in the High Performance Computing Center of Jilin University.
Author information
Authors and Affiliations
Contributions
Y.M. proposed the research. Y.W., H.L., J.L., L.Z., H.W. and Y.M. did the calculations and analysed the data. Y.W. and Y.M. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S4, Supplementary Table S1 and Supplementary Methods. (PDF 960 kb)
Rights and permissions
About this article
Cite this article
Wang, Y., Liu, H., Lv, J. et al. High pressure partially ionic phase of water ice. Nat Commun 2, 563 (2011). https://doi.org/10.1038/ncomms1566
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms1566
This article is cited by
-
Formation of ammonia–helium compounds at high pressure
Nature Communications (2020)
-
Room temperature electrofreezing of water yields a missing dense ice phase in the phase diagram
Nature Communications (2019)
-
Topologically frustrated ionisation in a water-ammonia ice mixture
Nature Communications (2017)
-
Materials discovery at high pressures
Nature Reviews Materials (2017)
-
Nonmetallization and band inversion in beryllium dicarbide at high pressure
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
