
Abstract
Feglymycin is a naturally occurring, anti-HIV and antimicrobial 13-mer peptide that includes highly racemizable 3,5-dihydroxyphenylglycines (Dpgs). Here we describe the total synthesis of feglymycin based on a linear/convergent hybrid approach. Our originally developed micro-flow amide bond formation enabled highly racemizable peptide chain elongation based on a linear approach that was previously considered impossible. Our developed approach will enable the practical preparation of biologically active oligopeptides that contain highly racemizable amino acids, which are attractive drug candidates.
Similar content being viewed by others
Introduction
Biologically active peptides have garnered considerable attention as drug candidates because their target specificity is generally higher than that of small molecule-based drugs, and their production cost is lower than that of protein-based drugs1,2,3. In particular, oligopeptides consisting of <20 amino acids account for three-fourths of existing marketed peptide drugs3. Therefore, efficient and practical synthesis of these oligopeptides is highly important. In general, a linear/convergent hybrid approach is used for the synthesis of these oligopeptides: (1) short peptides (<10 amino acids) are prepared based on a linear strategy wherein peptide chains are elongated one-by-one. This linear strategy enables the facile installation of various amino acids for rapid analogue synthesis, and it minimizes the protecting group manipulation of terminal carboxylic acids while avoiding undesired diketopiperazine formation; and (2) the prepared short peptides are coupled in a convergent manner to afford the target oligopeptides because longer peptides are often poorly soluble against solvents. In addition, this approach facilitates purification because the molecular weight of the desired oligopeptides is much different from that of their precursors.
Many arylglycine-containing biologically active oligopeptides have been reported, and they are highly important as drugs and drug candidates. Phenylglycine (Phg), 4-hydroxyphenylglycine (Hpg) and 3,5-dihydroxyphenylglycine (Dpg) are the most important representatives of the arylglycines because they can be found in various biologically active natural products such as formadicin, ramoplanin, vancomycin and teicoplanin4. These arylglycines significantly contribute to the biological activity of these natural products4.
Despite their importance, synthetic methodologies that can be used for arylglycines-containg oligopeptides is very limited due to their racemization-prone nature. Reportedly, phenylglycine is 60 times more prone to racemization than alanine5. Dpg is even more racemization-prone than the phenylglycine4.
Feglymycin isolated from Streptomyces sp. DSM 11171 (refs 6, 7) is a biologically active oligopeptide composed of 13 amino acids involving Hpgs and highly racemizable Dpgs. The biological activity of feglymycin involves strong anti-HIV activity8 and moderate antimicrobial activity9,10, and its unique helical conformation11 makes it an attractive lead compound for drug development. In 2009, the Süssmuth group reported the first total synthesis of feglymycin based on a highly convergent approach, in which D-Dpgs were coupled with the neighbouring C-terminal side of amino acids at an initial stage12. This amidation of the D-Dpgs with amino acids was performed using a coupling agent, 3-(diethyloxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT). Then, the resultant dipeptides/tripeptides were coupled to afford hexapeptide and heptapeptide. The DEPBT was crucial to avoid an undesired racemization reaction. A linear approach to the syntheses of the hexapeptide and the heptapeptide would have previously described merits. However, the Süssmuth group reported that the coupling of D-Dpgs with longer peptides based on a linear approach resulted in severe racemization of the D-Dpgs and provided an inseparable mixture of diastereomers even when using DEPBT. The Süssmuth group concluded that a linear approach for the hexapeptide and the heptapeptide containing D-Dpgs was not possible12. The development of peptide chain elongation based on a linear approach is important not only for the synthesis of feglymycin and related compounds, but also for the synthesis of other arylglycines-containg biologically active oligopeptides.
Recent marked progress in micro-flow technology13,14,15,16,17,18,19,20 has enabled precise control of the reaction time (<1 s) and temperature21,22,23. We have studied micro-flow acylations24,25 and recently reported micro-flow amide bond formation using a high atom economy and an inexpensive coupling agent, triphosgene26. We envisaged that if micro-flow amide bond formation enables the amidation of Dpgs without severe racemization, then it should be possible to use a linear synthetic approach to prepare the hexapeptide and the hepapeptide.
Herein, we wish to report the total synthesis of feglymycin based on a linear/convergent hybrid synthetic approach using a micro-flow amide bond formation that suppresses the undesired racemization. In detail, we planned to couple the hexapeptide and the heptapeptide in a convergent manner for the total synthesis of feglymycin and synthesize both oligo peptides based on a linear approach using micro-flow amide bond formation. The challenge was to suppress any undesired racemization of the highly racemizable Dpgs using our micro-flow amidation.
Results
Examination of micro-flow amide bond formation of arylglycines
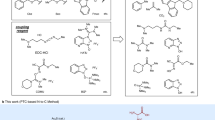
The key micro-flow amide bond formation of arylglycines was examined using readily available D-Hpgs 10 and 16 (Table 1). We connected two T-shaped mixers with Teflon tubing and immersed them in a water bath. A solution of carboxylate 10 in solvent A was introduced into the first mixer with a syringe pump. The solution of triphosgene in MeCN was also introduced into the first mixer with a syringe pump to rapidly generate symmetric anhydride in situ26 (<0.5 s). To accomplish amidation (<4.3 s), a solution of amine 16 in solvent B was then introduced into the second mixer with a syringe pump. The reaction was quenched by pouring the mixture into a saturated aqueous solution of NH4Cl, brine and ethyl acetate (micro-flow reactor setup, see Supplementary Methods). To investigate the influence of the protecting group on a nitrogen atom (=P), three protecting groups were examined (Table 1): t-butoxycarbonyl (Boc), 10a; benzyloxycarbonyl (Cbz), 10b; and, allyloxycarbonyl (Alloc), 10c. The 9-fluorenylmethoxycarbonyl (Fmoc) group was not examined because we feared that the basic deprotection conditions might cause undesired racemization. We did not protect the phenolic hydroxyl group (pKa=ca. 10) in D-Hpgs 10 because we did not expect the carboxylate generated from an equimolar amount of N,N-diisopropylethylamine (DIEA) and D-Hpg (pKa of CO2H in D-Hpg=ca. 2) to deprotonate the phenolic hydroxyl group in D-Hpg to afford an undesired highly nucleophilic phenoxide. On the other hand, the nucleophilicity of neutral phenolic hydroxyl group is much lower than that of an amino group, and, therefore, we speculated that the undesired nucleophilic attack of the phenolic hydroxyl group would not occur. In our previous report, N,N-dimethylformamide (DMF) and MeCN were used for solvents A and B, respectively26. However, carboxylate 10a and amine 16 were not soluble against these solvents. Therefore, N,N-dimethylacetoamide (DMA) was used instead of DMF and MeCN (entries 1–5). Carboxylates 10b (P=Cbz) and 10c (P=Alloc) afforded the desired products 18b and 18c in good yields (entries 2 and 3), respectively, whereas 10a (P=Boc) afforded 18a in a low yield (entry 1). To suppress racemization, the reaction was carried out at 10 °C. As expected, the generation of undesired epimers 19b and 19c was suppressed (entries 4 and 5), although a slight decrease in yield was observed in the case of carboxylate 10b (entry 2 versus 4). The use of H2O/MeCN for solvent B improved the yield of 18b in the case of carboxylate 10b (P=Cbz) without increasing racemization (entry 4 versus 6). On the other hand, in the case of 10c (P=Alloc), comparable results were observed (entry 5 versus 7). The corresponding batch reaction resulted in increased racemization (entry 7 versus 8). The use of N-methylpyrrolidone (NMP) and N,N′-dimethylpropyleneurea (DMPU) that could dissolve 10c resulted in a decrease in the yields (entries 9 and 10). Entries 5 (condition A) and 7 (condition B) were used for the coupling of arylglycines retaining an Alloc group, and entry 6 (condition C) was used for the coupling of arylglycine retaining a Cbz group in the following peptide chain elongations. We decided to use the Cbz group for P1 and the Alloc group for P2 (Fig. 1) because the Cbz group could be removed along with the benzyl (Bn) group under hydrogenolysis conditions.

Our linear synthetic approach and Süssmuth’s convergent synthetic approaches. Süssmuth concluded that the linear approach for oligopeptides 2 and 3 containing D-Dpgs was not possible.
Synthesis of C-terminal hexapeptide 2
We started with a synthesis of the C-terminal dipeptide 20 (Fig. 2). Micro-flow amide bond formation afforded a mixture of 5, 20 and the HCl salt of DIEA. The desired dipeptide 20 was readily isolated via simple aqueous workup and recrystallization (70% yield, 13 g). Salt-free carboxylic acid 5 was also recovered in a pure form (63% based on unreacted 5) via simple aqueous workup and recrystallization.

Dipeptide 20 was prepared by micro-flow amide bond formation. The obtained dipeptide was readily purified via simple aqueous workup and recrystallization.
Peptide chain elongation was performed based on the linear approach, as shown in Fig. 3. First, an Alloc group was removed from the protected dipeptide 20 under neutral conditions27,28,29 using a batch reactor. Micro-flow amidation with carboxylate 6 was performed under condition A with slight modification (activation time: 1.0 s), because dipeptide 11 was not dissolved in the H2O/MeCN mixed solvent with the use of condition B. After recrystallization, the desired pure tripeptide 21, which did not contain an epimer, was obtained in a 73% yield (two steps). To our delight, a coupling of 22 with highly racemizable D-Dpg 7 (refs 30, 31) afforded the desired pure tetrapeptide 23, with no epimer, in a 79% yield (two steps) under condition A. The coupling of sterically hindered carboxylate 8 and tetrapeptide 24 afforded the desired pure pentapeptide 25 in a 51% yield (two steps). The removal of the Alloc group of protected pentapeptide 25 was performed using a solid-immobilized Pd catalyst in a batch reactor to facilitate the purification of polar pentapeptide. The subsequent coupling of highly racemizable D-Dpg 7 with pentapeptide 26 afforded the desired pure hexapeptide 2 with no epimer in a 70% yield (two steps).

Conditions: (a) Pd(PPh3)4, PhSiH3, CH2Cl2/MeOH, r.t., 1–1.5 h, batch. (b) PS-Ph3P-Pd, PhSiH3, CH2Cl2/MeOH/H2O, r.t., 1 h, batch.
Synthesis of N-terminal heptapeptide 3
Synthesis of N-terminal heptapeptide 3 was performed based on the linear approach, as shown in Fig. 4. The coupling of 9 with highly racemizable D-Dpg 7 under condition A caused a substantial generation of the undesired epimer (11%), whereas condition B afforded the desired peptide 27 in a 76% yield without severe racemization (1%). Dipeptide 14 was coupled with 6 under condition B to afford the desired tripeptide 28 in a 66% yield (two steps). The coupling of 29 with highly racemizable D-Dpg 7 was performed under condition B to afford tetrapeptide 30 in a 49% yield (two steps). The next coupling reaction of tetrapeptide 31 with 8 was performed to afford pentapeptide 32 in a 36% yield (two steps). In this reaction, 31% of the unreacted tetrapeptide 31 was recovered. The subsequent coupling of 33 with highly racemizable D-Dpg 7 under condition B using 5.0 equiv. of 7, and 0.8 equiv. of triphosgene afforded the desired hexapeptide 34 in a 51% yield (two steps). The use of an excess amount of 7 and triphosgene was important for the return of a good yield. In this reaction, 34% of unreacted pentapeptide 33 was recovered. Hexapeptide 35 was coupled with 10b under condition B using 5.0 equiv. of 10b and 0.8 equiv. of triphosgene, and the subsequent preparative thin layer chromatography separation afforded the pure heptapeptide 3 with no epimer in a 25% yield (two steps). In this reaction, 24% of the unreacted 35 was recovered. In these reactions as well as in other amide bond formations, expensive Hpgs and Dpgs were recovered and reused. It should be noted that peptide chain elongation without severe racemization was achieved based on a linear approach that was considered impossible in feglymycin total synthesis. In addition, the coupling of racemizable amino acids usually requires low temperature and a long reaction time, whereas our developed process allowed rapid coupling (≤5.3 s).

Conditions: (a) Pd(PPh3)4, PhSiH3, CH2Cl2/MeOH/H2O, r.t., 40 min, batch. (b) PS-Ph3P-Pd, PhSiH3, CH2Cl2/MeOH/H2O, r.t., 1.5–2.5 h, batch.
Total synthesis of feglymycin (1)
Deprotection and coupling of hexapeptide 2 and heptapeptide 3 was performed in accordance with the Süssmuth group report (Fig. 5). The structure of obtained feglymycin (1) was confirmed by 1H and 13C NMR, IR, and HRMS spectra, as well as by specific rotation. The observed spectra were consistent with the previously reported data12.

Conditions: (a) Me3SnOH, 1,2-dichloroethane; 85 °C, 3.5 h; (b) PS-Ph3P-Pd, PhSiH3, CH2Cl2/MeOH/H2O, r.t., 50 min; (c) DEPBT, NaHCO3, DMF, 0 °C, 24 h, then 25 °C, 11 h, 2 steps 44% from heptapeptide 3. (d) H2, Pd/C, MeOH, r.t., 3 h, quant.
Discussion
In summary, we demonstrated a total synthesis of feglymycin based on a linear/convergent hybrid synthetic approach. Our originally developed micro-flow amide bond formation enabled the efficient preparation of hexapeptide 2 and heptapeptide 3 containing highly racemizable D-Dpgs based on a linear synthetic approach that was previously thought to be impossible. The developed synthetic approach will be useful for the rapid preparation of feglymycin analogues in the future. Our micro-flow amide bond formation uses triphosgene, and only emits CO2 and HCl salt of DIEA. One of the advantages of using microreactors is the ease of scaling up. It should be possible to scale-up our developed process by either continuous operation or by a numbering-up of the microreactors. Our developed process will enable the practical preparation of biologically active oligopeptides containing highly racemizable amino acids.
Methods
General
NMR spectra were recorded on JEOL Model ECP-400 (400 MHz for 1H, 100 MHz for 13C) or Bruker Biospin AVANCE II 400 (400 MHz for 1H, 100 MHz for 13C), Bruker Biospin AVANCE III HD 500 (500 MHz for 1H, 125 MHz for 13C) instrument in the indicated solvent. Chemical shifts are reported in units of parts per million (p.p.m.) relative to the signal (0.00 p.p.m.) for internal tetramethylsilane for solutions in CDCl3 (7.26 p.p.m. for 1H, 77.0 p.p.m. for 13C) or DMSO (2.50 p.p.m. for 1H, 39.5 p.p.m. for 13C). Multiplicities are reported by using the following abbreviations: s; singlet, d; doublet, t; triplet, q; quartet, m; multiplet, br; broad, J; coupling constants in Hertz (Hz). IR spectra were recorded on Perkin-Elmer Spectrum One FT-IR spectrometer or JASCO FT/IR-4100. Only the strongest and/or structurally important peaks are reported as IR data given in cm−1. Optical rotations were measured using a JASCO P-1020 or P-2200, Rudolph Research Analytical AUTOPOL IV. HRMS (ESI-TOF) were measured with a Bruker micrOTOF II.
All reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60F-254) with ultraviolet light, visualized by 10% ethanolic phosphomolybdic acid or 0.5% ninhydrin n-butanol solution. Flash column chromatography was performed on Silica Gel 60 N purchased from Kanto Chemical Co or Silica Gel PSQ 60B purchased from Fuji Silysia Chemical LTD. Analytical HPLC was carried out on Shimadzu LC-10AT VP Liquid Chromatograph with a Shimadzu RID-10A Refractive Index Detector and a Shimadzu SPD-10A VP UV–vis Detector, Shimadzu SCL-10A VP System Controller. MeCN was dried using a Glass Contour. DIEA was distilled from ninhydrin and KOH. For HPLC analysis of 18a–c and 19a–c, see Supplementary Figs 2–7, and for NMR spectra of synthesized compounds and analysis of 18a–c and 19a–c, see Supplementary Figs 8–45.
Data availability
The authors declare that the data supporting the findings of this study are available within the article (and its Supplementary Information files). The data supporting the findings of this study are available from the authors upon reasonable request.
Additional information
How to cite this article: Fuse, S. et al. Total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation. Nat. Commun. 7, 13491 doi: 10.1038/ncomms13491 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Neefjes, J. & Ovaa, H. A peptide’s perspective on antigen presentation to the immune system. Nat. Chem. Biol. 9, 769–775 (2013).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122–128 (2015).
Craik, D. J., Fairlie, D. P., Liras, S. & Price, D. The future of peptide-based drugs. Chem. Biol. Drug Design 81, 136–147 (2013).
Al Toma, R. S., Brieke, C., Cryle, M. J. & Süssmuth, R. D. Structural aspects of phenylglycines, their biosynthesis and occurrence in peptide natural products. Nat. Prod. Rep. 32, 1207–1235 (2015).
Smith, G. G. & Sivakua, T. Mechanism of the racemization of amino acids. Kinetics of racemization of arylglycines. J. Org. Chem. 48, 627–634 (1983).
Vértesy, L. et al. New antibiotic, feglymycine, process for its preparation and use of the same. European patent EP-B1 0848064 (1998).
Vértesy, L. et al. Feglymycin, a novel inhibitor of the replication of the humanimmunodeficiency virus. J. Antibiot. 52, 374–382 (1999).
Férir, G. et al. Feglymycin, a unique natural bacterial antibiotic peptide, inhibits HIV entry by targeting the viral envelope protein gp120. Virology 433, 308–319 (2012).
Rausch, S. et al. Feglymycin is an inhibitor of the enzymes MurA and MurC of the peptidoglycan biosynthesis pathway. Chembiochem 12, 1171–1173 (2011).
Hänchen, A. et al. Alanine scan of the peptide antibiotic feglymycin: assessment of amino acid side chains contributing to antimicrobial activity. Chembiochem 14, 625–632 (2013).
Bunkóczi, G., Vértesy, L. & Sheldrick, G. M. The antiviral antibiotic feglymycin: first direct-methods solution of a 1000+ equal-atom structure. Angew. Chem. Int. Ed. 44, 1340–1342 (2005).
Dettner, F. et al. Total synthesis of the antiviral peptide antibiotic feglymycin. Angew. Chem. Int. Ed. 48, 1856–1861 (2009).
Watts, P., Wiles, C., Haswell, S. J., Pombo-Villar, E. & Styring, P. The synthesis of peptides using micro reactors. Chem. Commun. 2001, 990–991 (2001).
Watts, P., Wiles, C., Haswell, S. J. & Pombo-Villar, E. Solution phase synthesis of beta-peptides using micro reactors. Tetrahedron 58, 5427–5439 (2002).
Watts, P., Wiles, C., Haswell, S. J. & Pombo-Villar, E. Investigation of racemisation in peptide synthesis within a micro reactor. Lab. Chip. 2, 141–144 (2002).
France, S., Bernstein, D., Weatherwax, A. & Lectka, T. Performing the synthesis of a complex molecule on sequentially linked columns: toward the development of a ‘synthesis machine’. Org. Lett. 7, 3009–3012 (2005).
Baxendale, I. R., Ley, S. V., Smith, C. D. & Tranmer, G. K. A flow reactor process for the synthesis of peptides utilizing immobilized reagents, scavengers and catch and release protocols. Chem. Commun. 4835–4837 (2006).
Flögel, O., Codée, J. D. C., Seebach, D. & Seeberger, P. H. Microreactor synthesis of β-peptides. Angew. Chem. Int. Ed. 45, 7000–7003 (2006).
Zhang, Y., Blackman, M. L., Leduc, A. B. & Jamison, T. F. Peptide fragment coupling using a continuous-flow photochemical rearrangement of nitrones. Angew. Chem. Int. Ed. 52, 4251–4255 (2013).
Ramesh, S. et al. Microreactors for peptide synthesis: looking through the eyes of twenty first century !!!. Amino Acids 46, 2091–2104 (2014).
Yoshida, J.-I. Flash Chemistry: Fast Organic Synthesis in Microsystems WILEY-VCH (2008).
Yoshida, J.-I. Flash chemistry: flow microreactor synthesis based on high-resolution reaction time control. Chem. Rec. 10, 332–341 (2010).
Yoshida, J.-I., Nagaki, A. & Yamada, T. Flash chemistry: fast chemical synthesis by using microreactors. Chem. Eur. J. 14, 7450–7459 (2008).
Fuse, S., Tanabe, N. & Takahashi, T. Continuous in situ generation and reaction of phosgene in a microflow system. Chem. Commun. 47, 12661–12663 (2011).
Fuse, S., Tanabe, N., Tannna, A., Konishi, Y. & Takahashi, T. Micro-flow synthesis and structural analysis of sterically crowded diimine ligands with five aryl rings. Beilstein. J. Org. Chem. 9, 2336–2343 (2013).
Fuse, S., Mifune, Y. & Takahashi, T. Efficient amide bond formation through a rapid and strong activation of carboxylic acids in a microflow reactor. Angew. Chem. Int. Ed. 53, 851–855 (2014).
Dessolin, M., Guillerez, M.-G., Thieriet, N., Guibé, F. & Loffet, A. New allyl group acceptors for palladium catalyzed removal of allylic protections and transacylation of allyl carbamates. Tetrahedron Lett. 36, 5741–5744 (1995).
Thieriet, N., Alsina, J., Giralt, E., Guibé, F. & Albericio, F. Use of Alloc-amino acids in solid-phase peptide synthesis. Tandem deprotection-coupling reactions using neutral conditions. Tetrahedron Lett. 38, 7275–7278 (1997).
Bo, L. i. D. & Robinson, J. A. An improved solid-phase methodology for the synthesis of putative hexa- and heptapeptide intermediates in vancomycin biosynthesis. Org. Biomol. Chem. 3, 1233–1239 (2005).
Mifune, Y., Fuse, S. & Tanaka, H. Synthesis of N-allyloxycarbonyl 3,5-dihydroxyphenylglycine via photochemical Wolff rearrangement–nucleophilic addition sequence in a micro-flow reactor. J. Flow Chem. 4, 173–179 (2014).
Fuse, S., Otake, Y., Mifue, Y. & Tanaka, H. A facile preparation of α-aryl carboxylic acid via one-flow Arndt-Eistert synthesis. Aust. J. Chem. 68, 1657–1661 (2015).
Acknowledgements
This work was partially supported by a Grant-in-Aid for Young Scientists (B), Scientific Research on Innovative Areas 2707 Middle molecular strategy form MEXT (no. 15H05849), and The Naito Foundation Natural Science Scholarship.
Author information
Authors and Affiliations
Contributions
S.F. and Y.M. conceived and designed project and wrote the paper. Y.M. performed all experiments by himself. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-45 and Supplementary Methods. (PDF 5338 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Fuse, S., Mifune, Y., Nakamura, H. et al. Total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation. Nat Commun 7, 13491 (2016). https://doi.org/10.1038/ncomms13491
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms13491
This article is cited by
-
Verification of preparations of (1H-indol-3-yl)methyl electrophiles and development of their microflow rapid generation and substitution
Communications Chemistry (2023)
-
Arylglycine: A Focus on Amino Acid Preparation and Peptide Synthesis
International Journal of Peptide Research and Therapeutics (2022)
-
Challenges and outlook for catalytic direct amidation reactions
Nature Catalysis (2019)
-
Accelerated microfluidic native chemical ligation at difficult amino acids toward cyclic peptides
Nature Communications (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
