
Abstract
The activity of electrocatalysts exhibits a strongly dependence on their electronic structures. Specifically, for perovskite oxides, Shao-Horn and co-workers have reported a correlation between the oxygen evolution reaction activity and the eg orbital occupation of transition-metal ions, which provides guidelines for the design of highly active catalysts. Here we demonstrate a facile method to engineer the eg filling of perovskite cobaltite LaCoO3 for improving the oxygen evolution reaction activity. By reducing the particle size to ∼80 nm, the eg filling of cobalt ions is successfully increased from unity to near the optimal configuration of 1.2 expected by Shao-Horn’s principle. Consequently, the activity is significantly enhanced, comparable to those of recently reported cobalt oxides with eg∼1.2 configurations. This enhancement is ascribed to the emergence of spin-state transition from low-spin to high-spin states for cobalt ions at the surface of the nanoparticles, leading to more active sites with increased reactivity.
Similar content being viewed by others


Introduction
Electrochemical water splitting is regarded as a prime approach for renewable energy conversion and storage1,2. One of the critical reactions of this process is oxygen evolution reaction (OER). However, this reaction is kinetically sluggish due to a complex multistep four-electron oxidation1,2,3,4,5. To reach a desirable current density of 10 mA cm−2, which is a metric associated with solar fuel synthesis, a considerable overpotential (η) relative to thermodynamic potential of the reaction is required, but will hinder the large-scale electrochemical water splitting3,4. Currently, RuO2 and IrO2 are among the most active OER catalysts, however, their unacceptable cost and low abundance severely restrict their large-scale applications5,6. Therefore, it is of great technological and scientific significance to pursue highly efficient alternatives on the basis of earth-abundant nonprecious materials.
Transition-metal oxides and their derivatives have received much attention because of their earth-abundant reserves, low cost, environment-friendly features and remarkable OER activities7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23. Specially, cobalt oxides, such as layered LiCoO2 (refs 11, 12), Co-oxyhydroxides (refs 13, 14), spinel Co3O4 (refs 15, 16, 17), perovskite Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF)18 and PrBaCo2O5+δ (ref. 19), have been explored as potential OER catalysts due to their high activities comparable to and even better than the precious metal oxides. Theoretical and experimental works have shown that the OER activity is intrinsically related to the electronic structure of Co ions, including oxidation state and spin configuration, which enlightens the rational design of optimal catalysts7,8,9,10,11,12,13,14,15,16,17,18,19,20,21. For example, Lu et al.12 recently reported that the OER activity of LiCoO2 was remarkably enhanced by tuning the Co oxidation state via electrochemical delithiation process. More interestingly, for perovskite transition-metal oxides, Shao-Horn and co-workers18 demonstrated the direct dependence of OER activity on the eg orbital filling of the transition-metal ions, and that the oxide BSCF with eg∼1.2 configuration exhibited the highest OER activity. The performance of the BSCF was found to be nearly identical to IrO2 in terms of catalytic activity. However, this Co-based oxide undergoes surface amorphization after long-term potential cycles under OER conditions22. Therefore, substantial progress is still needed to develop perovskite-type OER catalysts with improved activity and stability. In this respect, Shao-Horn’s principle highlights that the optimization of eg filling close to 1.2 should be an alternative strategy to develop the transition-metal oxides as the effective OER catalysts. Practically, a recent study by Zhu et al.23 supported this principle, where an improved OER activity was realized through adjusting the eg filling to ∼1.2 with partial substitution of Co by Nb in SrCo0.8Fe0.2O3−δ (ref. 23).
In this work, we present a facile method to modify the electronic structure of Co ions by varying the particle size for the improvement in the OER activity. We focus on the perovskite cobaltite LaCoO3 (LCO), which is well-known for its unique thermally-driven transition of Co3+ ions from low spin (LS: t2g6eg0) at low temperatures to higher spin state with eg orbital configuration of eg∼1.0 at room temperature18,24,25,26,27,28,29,30,31. This compound was reported to exhibit reasonable OER activity but much less than BSCF18. Here, by reducing the particle size, the eg filling is successfully increased from unity to close to the optimization configuration of ∼1.2. As a consequence, the OER activity of the 80-nm LCO is higher than those of other sized samples as well as the bulk, and comparable to those of the reported cobalt oxides with eg∼1.2 filling, which enables the nanosized LCO to be applicable as a promising OER catalyst.
Results
Crystal structures of the bulk and nanosized LCO
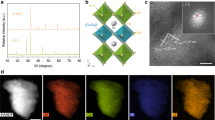
The LCO samples were prepared by a sol–gel method32,33. The precursory powders derived from the gel were annealed at 600, 700 and 800 °C for 6 h to produce the LCO nanoparticles with the particle size of about 60, 80 and 200 nm (ref. 32), respectively, as well as at 1,000 °C for 12 h to the bulk sample with the particle size of about 0.5–1 μm (Supplementary Fig. 1). A representative transmission electron microscopy (TEM) image for the 700 °C annealing sample is shown in Fig. 1a. The high-resolution TEM images and the selective area electron diffraction patterns reveal a single-crystal structure of small LCO particles with a high crystallinity (Supplementary Fig. 2). The X-ray diffraction patterns (Fig. 1b and Supplementary Fig. 3) reveal that all the samples take a rhombohedral structure with R  c space group (Fig. 1c). The structural parameters obtained from the Rietveld refinements on the diffraction data are given in Supplementary Table 1. As the particle size is reduced, the unit cell is found to be expanded. Specially, the bond length of Co–O exhibits an obvious increase (Fig. 1d).
c space group (Fig. 1c). The structural parameters obtained from the Rietveld refinements on the diffraction data are given in Supplementary Table 1. As the particle size is reduced, the unit cell is found to be expanded. Specially, the bond length of Co–O exhibits an obvious increase (Fig. 1d).

(a) TEM image for the 80 nm LCO. (b) X-ray diffraction patterns for bulk and 80 nm LCO together with the Rietveld refined results. (c) LCO crystal structure. (d) The length of Co–O bond for the bulk and nanosized LCO. Scale bar, 180 nm.
Spin structures of the bulk and nanosized LCO
The temperature-dependent magnetizations were measured with a magnetic field of H=1 kOe under field-cooling procedures for all the samples (Supplementary Fig. 4a) to study the spin structures of Co ions controlled by the particle size. Above 150 K, the susceptibilities derived from the magnetizations (χ=M/H) obey a paramagnetic Curie–Weiss law:  , where C is Curie constant, and Θ is Curie–Weiss temperature. From the fitting results (Fig. 2a), an effective magnetic moment μeff can be calculated through
, where C is Curie constant, and Θ is Curie–Weiss temperature. From the fitting results (Fig. 2a), an effective magnetic moment μeff can be calculated through  μB (Supplementary Fig. 4b). For the bulk LCO, although the exact nature of whether the higher spin state of Co3+ ions at room temperature is intermediate-spin (IS: t2g5eg1) state or a mixture of LS and high spin (HS: t2g4eg2) states was controversial in the past decades24,25,26,27, a large number of recent theoretical and experimental studies reveal that the mixture of LS and HS states is more favourable28,29,30,31. Here, the calculated μeff of 3.48 μB for the bulk sample is well consistent with those values reported for the polycrystalline bulk LCO, which corresponds to the Co3+ ions in 50% HS+50% LS states as well as the eg filling of ∼1.0 (refs 25, 28, 29, 30, 31). For the nanosized LCO, μeff shows a gradual increase with the decrease of the particle size, suggesting that the spin state of partial Co ions transmits from LS to HS state. Using the calculated μeff, the spin states are estimated to be 55% HS+45% LS, 60.5% HS+39.5% LS and 63.7% HS+36.3% LS for the 200, 80 and 60 nm samples (Supplementary Note 1 and Supplementary Table 2), meaning that about 5, 10.5 and 13.7% Co3+ ions in LS state change to be in HS state, respectively. As shown in Fig. 2b, the corresponding eg filling is about 1.1, 1.2 and 1.27 for the nanosized LCO, respectively. It is worthwhile to emphasize that by reducing the particle size to about 80 nm, we successfully tune the eg filling from unity to the optimization value of 1.2 expected by Shao-Horn’s principle.
μB (Supplementary Fig. 4b). For the bulk LCO, although the exact nature of whether the higher spin state of Co3+ ions at room temperature is intermediate-spin (IS: t2g5eg1) state or a mixture of LS and high spin (HS: t2g4eg2) states was controversial in the past decades24,25,26,27, a large number of recent theoretical and experimental studies reveal that the mixture of LS and HS states is more favourable28,29,30,31. Here, the calculated μeff of 3.48 μB for the bulk sample is well consistent with those values reported for the polycrystalline bulk LCO, which corresponds to the Co3+ ions in 50% HS+50% LS states as well as the eg filling of ∼1.0 (refs 25, 28, 29, 30, 31). For the nanosized LCO, μeff shows a gradual increase with the decrease of the particle size, suggesting that the spin state of partial Co ions transmits from LS to HS state. Using the calculated μeff, the spin states are estimated to be 55% HS+45% LS, 60.5% HS+39.5% LS and 63.7% HS+36.3% LS for the 200, 80 and 60 nm samples (Supplementary Note 1 and Supplementary Table 2), meaning that about 5, 10.5 and 13.7% Co3+ ions in LS state change to be in HS state, respectively. As shown in Fig. 2b, the corresponding eg filling is about 1.1, 1.2 and 1.27 for the nanosized LCO, respectively. It is worthwhile to emphasize that by reducing the particle size to about 80 nm, we successfully tune the eg filling from unity to the optimization value of 1.2 expected by Shao-Horn’s principle.

(a) The temperature dependence inverse susceptibilities for all the LCO samples. The dotted lines are the fitting results by a Curie–Weiss law. (b) The corresponding eg filling. (c,d) Representative EELS spectra of the 80 nm LCO at Co L-edge and O K-edge, respectively. The inset corresponds to the representative position of EELS acquisition. Scale bar, 50 nm.
To further confirm the spin-state transition and to explore its possible origin, the electron energy loss spectroscopy (EELS) analyses on the LCO nanoparticles were performed on the scanning TEM. Figure 2c,d shows the representative EELS spectra at Co L- and O K-edges from the centre and edge of the 80 nm nanoparticle, which are sensitive to the electronic structure from the core (bulk) and shell (surface), respectively34,35. For the Co L-edge spectra, no noticeable changes in the L3/L2 ratio between the bulk and surface were found, suggesting that the oxidation state of Co ions remained unchanged35,36,37,38,39. For the O K-edge spectra, three characteristic peaks near the edge onset, labelled a, b and c, were observed. The prepeak a, b and c were assigned to the hybridization of O 2p with Co 3d, La 5d and Co 4sp orbitals, respectively36,37,38,39. Compared with that from the centre position, the spectrum from the edge shows an obvious reduction in the intensity of the prepeak a. Similar results are also found in the other nanoparticles (Supplementary Fig. 5). This reduction is generally attributed to the formation of oxygen vacancy or the weakening of Co 3d–O 2p hybridization36,37,38,39. Since the Co L-edge spectra revealed no change in the Co oxidation state, the formation of oxygen vacancies can be excluded. Thus, the modification of O K-edge would originate from the change of the Co 3d–O 2p hybridization. Previous works have been widely reported that the spin-state transition of Co3+ ions in the LCO can significantly modify the hybridization of Co 3d–O 2p orbitals and then the intensity of the prepeak a (refs 35, 36, 37, 38, 39). In the LS configuration, the 3d eg levels of Co3+ ions are completely empty, allowing the electrons from the filled O 2p levels to be shared with Co eg orbitals, and accordingly creating O 2p holes. Thus, the hybridization of Co 3d with the O 2p states promotes electron transitions between 1s and the unfilled O 2p state, resulting in the prepeak a of the O K-edge. However, as a HS state of the Co3+ ions emerges, the eg orbitals are increasingly occupied, which prevents the charge transfer and weakens the hybridization of O 2p with Co 3d orbitals, resulting in the decreased intensity of the prepeak a (refs 36, 37, 38, 39). Therefore, the decrease in the intensity of the prepeak a at the surface confirms the existence of surface spin-state transitions in the nanosized LCO.
We proposed a mechanism to explain the presence of surface spin-state transitions of Co3+ ions where the modified crystal field splitting of Co 3d orbital at the surface favors the Co3+ ions to be in HS states, which has also been reported in nanosized stoichiometric LiCoO2 (ref. 40). Assuming that Co3+ ions within surface layers are all transited to be in HS state, we can give a rough estimate of the eg filling for the nanosized LCO on the basis of a simple core-shell model (Supplementary Note 2 and Supplementary Fig. 6). For nanosized perovskite oxides with the particle size ranging from tens to hundreds of nanometres, the surface layers are usually reported to be about 2–5 nm in the thickness41,42,43,44. Taking the thickness of 3 nm (see Supplementary Fig. 2), the estimated volume fractions of the surface layer are about 8.7, 20.8 and 27.1% for the 200, 80 and 60 nm LCO, respectively. Consequently, the increased fractions of the HS Co3+ ions are 4.4, 10.4 and 13.6%, which means that the eg fillings are increased to be about 1.09, 1.21 and 1.27, respectively. These values are well consistent with those obtained from the magnetizations, further supporting that the tuning of eg filling by the size reduction originates from the surface spin-state transition. In addition, since the radii of Co3+ ions increases when their spin state changes from LS to HS, the presence of this transition is also confirmed by the crystal structure data, where the expansion of the unit cell and the increase of Co–O length under decreasing the particle size are found (Supplementary Table 1 and Fig. 1d).
OER activities of the bulk and nanosized LCO
To shed light on the role that the surface spin-state transition plays in the OER activity for the LCO, the electrochemical measurements were carried out in O2-saturated 0.1 M KOH solutions using a standard three-electrode system. Figure 3a shows the iR-corrected polarization curves for all the samples, where the LCO nanoparticles exhibited smaller onset overpotentials than that of the bulk (∼0.37 V). In particular, the smallest onset overpotential of ∼0.32 V was observed in the 80 nm LCO. Similarly, the overpotential η required to achieve a current density of 10 mA cm−2 was also reduced from 0.62 V for the bulk to 0.54, 0.49 and 0.55 V as the particle size decreased to about 200, 80 and 60 nm, respectively. Figure 3b plots the dependence of mass activity at η=0.49 V on the eg filling. As the eg filling approached the optimal configuration of ∼1.2, the current density reached the largest value, which was about 4.3 times larger than that of the bulk. Moreover, the corresponding Tafel plots (Fig. 3d) also reveal that the 80 nm LCO possessed the smallest Tafel slope of ∼69 mV dec−1, much smaller than that of the bulk (∼102 mV dec−1). This large reduction suggests that the rate-determining step tends to change from the –OH adsorption to the O–OH formation18,45,46. In addition, the preliminary stability tests for the bulk and 80 nm LCO under a constant galvanostatic current of 10 A g−1 (Supplementary Fig. 7) also demonstrate that the 80 nm sample exhibited better stability than the bulk. As revealed by Co 2p X-ray photoelectron spectra of the 80 nm LCO before and after the electrolysis (Supplementary Fig. 8), no visible changes for the Co 2p spectra were found during the electrolysis, suggesting that the electronic state of the Co ions may remain unchanged. All of the above results clearly indicate that the OER activity of the LCO is successfully modified by controlling the particle size. As the particle size is reduced to about 80 nm with the eg∼1.2 configuration, the activity is significantly enhanced.

(a) Polarization curves of the bulk and nanosized LCO. (b) Mass and (c) special activities at η=0.49 V. (d) Tafel plots for the bulk and nanosized LCO. (e) Nyquist plots for the bulk and nanosized LCO. Error bars represent the s.d. from at least three independent measurements.
The origin of the enhanced OER activity
As the sample size is reduced, the enhanced mass catalytic activity towards the OER is largely ascribed to the increase of the surface area. Such scenarios were reported in various Co-based oxides such as BSCF18, SrNd0.1Co0.7Fe0.2O3−δ (ref. 23) and NiCo2O4 (ref. 47). However, in those cases, the size reduction leaded to a large decrease in the specific OER activities, that is, the normalized activities by the surface area. Since the specific activity reflects the intrinsic activity of the catalysis, this decrease indicates that intrinsic OER activity was deteriorated by reducing the sample size for those oxides. To clarify whether the enhancement of the OER activity in our LCO nanoparticles is intrinsic, the specific activities are further calculated on the basis of two types of surface areas, the Brunauer-Emmett-Teller (BET) surface areas and the electrochemically active surface areas, obtained by means of the gas desorption (Supplementary Fig. 9) and electrochemical double-layer capacitance measurements (Supplementary Fig. 10 and Supplementary Note 3), respectively. The specific activities at η=0.49 V normalized by the surface areas exhibit similar dependences on the eg filling to the mass activity (Supplementary Fig. 11). Compared with the bulk, the 80 nm sample is still 1.8 times more active in the specific activity normalized by the BET area (Fig. 3c), which strongly suggests that the increased number of active sites from the surface areas may be not the main contribution to the significant enhancement of the OER activity. The improved performance would be mainly attributed to the increased reactivity of the active sites due to the spin-state transition of Co3+ ions at surfaces. When the eg filling of Co3+ ions increases from about 1.0 to 1.2, the electron occupancy of the Co 3d–O 2p σ* band increases with the elongation of the length of Co–O bond as shown in Figs 1d and 2d. Thus, the hybridization of Co 3d–O 2p orbitals and the strength of Co–O bond become weaker, which leads to a less surface coverage by –OH groups on the active sites and thereby facilitates the formation of –OOH species45,46. As a result, the Tafel slope is reduced and the OER activity is improved. On the other hand, it has been generally demonstrated that H2O molecules are initially adsorbed onto the surface of catalysts during the OER process13,18,45. Consequently, the adsorption energy of H2O onto the active site plays a crucial role in the OER activity. Our density functional theory (DFT) calculations on the adsorption energy of H2O onto the surface Co ions in different spin states reveal that the surface Co ions being in HS state are more favourable for adsorbing H2O molecular (Supplementary Fig. 12 and Supplementary Table 3), well consistent with the improvement of the OER activity by reducing the particle size to 80 nm. However, it is worthwhile to note that as the particle size is further reduced to about 60 nm the activity decreases again, which cannot be explained by the above factors. We propose that the excessive eg occupancy (>1.2) of the Co3+ ions in this sample would make the charge transfer ability lower. As such, when the two neighboured Co ions in Co–O–Co network are both in HS state, the half-filling of eg orbitals tends to prevent the charge transfer. To confirm this point, the electrochemical impedance spectroscopy experiments have been carried out. As shown in Fig. 3e, the Nyquist plots reveal that the charge transfer resistance gradually decreases with the reduction of the particle size to 80 nm, while increases again as the size is further reduced to 60 nm. Therefore, we conclude that the modifications of the Co–O binding strength and the charge transfer ability associated with the surface spin-state transition in the LCO are responsible for the size-dependent OER activity.
Finally, we compared the OER activity of our LCO samples with those of the recently reported Co-based perovskite oxides with the optimal configuration of eg∼1.2. As illustrated in Table 1, it is interesting to find that the 80 nm LCO exhibits a well comparable activity with those well-known catalysts, which further consolidates Shao-Horn’s principle and suggests that tuning the spin state can provide an effective strategy to improve the OER activity.
Discussion
In summary, we highlight an effective strategy to engineer the electronic configuration of perovskite cobalt oxide for the development of high active electrocatalysts. By reducing the particle size to about 80 nm, the spin filling of Co ions in LCO is successfully tuned from unity (bulk) to near the optimization configuration of ∼1.2 expected by Shao-Horn’s principle. Through X-ray diffraction, magnetic measurements and EELS analysis, we confirm that this modification originates from the size-induced spin-state transition of Co3+ ions from LS to HS state. Consequently, the nanosized sample exhibits an improved OER activity with lower overpotential, smaller Tafel slope and better stability compared with the bulk. More interestingly, the performance of the 80 nm LCO can be comparable with those of the reported cobalt oxides with eg∼1.2 filling, suggesting that the LCO in this nanosized form can serve as a promising OER catalyst. Our work paves the way for the rational design of high-efficient OER catalysts.
Methods
Synthesis and characterization
La(NO3)3·6H2O and Co(NO3)2·6H2O were dissolved in deionized water, followed by the addition of a mixture of citric acid and ethylene glycol. Subsequently, the obtained transparent solution was slowly evaporated to get a gel, which was decomposed at about 400 °C for 4 h to result in dark brown powders. The precursor powders were further annealed at 600, 700 and 800 °C for 6 h to produce LCO nanoparticles with different particle sizes, and at 1,000 °C for 12 h to the bulk sample. The phase purity and crystal structure of the samples were determined by X-ray diffraction at room temperature on a Rigaku TTR-III diffractometer using Cu Ka radiation (λ=1.5418 Å). The field emission SEM and TEM images were obtained on a JEOL-2010 SEM and a JEM-2100F TEM, respectively. The HRTEM images and the EELS analyses were performed on a JEOL JEM-ARM200F TEM/scanning TEM with a spherical aberration corrector. The magnetic measurements were carried out with a MPMS SQUID magnetometer. The nitrogen adsorption−desorption isotherms were conducted on a Micromeritics ASAP 2000 system at 77 K. X-ray photoelectron spectra were carried out on an ESCALAB 250 X-ray photonelectron spectrometer with Al Kα as the excitation source.
Electrochemical measurements
The electrochemical tests were performed in O2-saturated 0.1 M KOH with a conventional three-electrode on the CHI660B electrochemical station. Saturated Ag/AgCl and platinum wires were used as the reference and the counter electrodes, respectively. The reference electrode was calibrated with respect to the reversible hydrogen electrode (RHE), which was carried out in the high-purity hydrogen saturated electrolyte with a Pt wire as the working electrode. Cyclic voltammetry was run at a sweep rate of 1 mV s−1. The average of the two potentials at which the current crossed zero was taken to be the thermodynamic potential for the hydrogen electrode reactions. In 0.1 M KOH, ERHE=EAg/AgCl+0.964 V. To prepare the working electrode, 3.5 mg of electrocatalyst and 20 μl of 5 wt% Nafion solutions were dispersed in 1 ml ethanol with sonication for at least 30 min to form a mixted ink. Then, 5 μl of this solution was drop-casted onto a 3 mm in diameter glassy carbon electrode and dried naturally, yielding a catalyst loading of 0.25 mg cm−2. Linear sweeping voltammograms were obtained at a scan rate of 5 mV s−1. The potentials are corrected to compensate for the effect of solution resistance, which were calculated by the following equation: EiR−corrected=E−iR, where i is the current, and R is the uncompensated ohmic electrolyte resistance (∼36 Ω) measured via high frequency ac impedance in O2-saturated 0.1 M KOH. The polarization curves were replotted as overpotential (η) vs log current (log j) to get Tafel plots for quantification of the OER activities of investigated catalysts. Electrochemical impedance spectroscopy were conducted with AC voltage with 5 mV amplitude at the potential of 1.67 V vs RHE within the frequency range from 100 KHz to 100 mHz. Durablity test was performed at room temperature under a constant galvanostatic current of 10 A g−1. Error bars represented s.d. from at least three independent measurements.
DFT calculations
DFT+U calculations with the Vienna ab initio simulation package48 for a water molecule adsorbed on (001) surface of LCO with LS and HS state on Co3+ ions were performed to study the influence on adsorption energy of water molecule with different spin states of Co3+ ions. A slab model consisting of eight atom-layers (La16Co16O48) was used to simulate the (001) surface with two terminations. In calculations, an effective U of 3.4 eV was added on Co 3d orbital, the plane wave energy cut-off was set to 400 eV, and a 2 × 2 × 1 Monkhorst–Pack k-point mesh was used. During geometry optimization, converge criteria were 10−5 eV for energy and 0.05 eV Å−1 for force. The LS of Co3+ was not obtained on CoO2 terminated (001) surface, since surface Co3+ was in a five-coordinated structure and turns into HS during the calculation even though it was set to LS initially.
Data availability
The data that support the findings of this study are available from the corresponding author on request.
Additional information
How to cite this article: Zhou, S. et al. Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition. Nat. Commun. 7:11510 doi: 10.1038/ncomms11510 (2016).
References
Gray, H. B. Powering the planet with solar fuel. Nat. Chem. 1, 7 (2009).
Lewis, N. S. & Nocera, D. G. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl Acad. Sci. USA 103, 15729–15735 (2007).
Cook, T. R. et al. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 110, 6474–6502 (2010).
Walter, M. G. et al. Solar water splitting cells. Chem. Rev. 110, 6446–6473 (2010).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Design of electrocatalysts for oxygen and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060–2086 (2015).
Lee, Y., Suntivich, J., May, K. J., Perry, E. E. & Shao-Horn, Y. Synthesis and activities of rutile IrO2 and RuO2 nanoparticles for oxygen evolution in acid and alkaline solutions. J. Phys. Chem. Lett. 3, 399–404 (2012).
Yuan, C. Z., Wu, H. B., Xie, Y. & Lou, X. W. Mixed transition metal oxides: design, controllable synthesis and energy-related applications. Angew. Chem. Int. Ed. 53, 1488–1504 (2014).
Wang, H. et al. Bifunctional non-noble metal oxide nanoparticle electrocatalysts through lithium-induced conversion for overall water splitting. Nat. Commun. 6, 7261 (2015).
Hong, W. T. et al. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 8, 1404–1427 (2015).
Kim, J. M., Yin, X., Tsao, K.-C., Fang, S. H. & Yang, H. A2B2O5 as oxygen deficient perovskite electrocatalyst for oxygen evolution reaction. J. Am. Chem. Soc. 136, 14646–14649 (2014).
Maiyalagan, T., Jarvis, K. A., Therese, S., Ferreira, P. J. & Manthiram, A. Spinel-type lithium cobalt oxide as a bifunctional electrocatalyst for the oxygen evolution and oxygen reduction reactions. Nat. Commun. 5, 3949 (2014).
Lu, Z. et al. Electrochemical tuning of layered lithium transition metal oxides for improvement of oxygen evolution reaction. Nat. Commun. 5, 5345 (2014).
Huang, J. H. et al. CoOOH nanosheets with high mass activity for water oxidation. Angew. Chem. Int. Ed. 54, 8722–8727 (2015).
Song, F. & Hu, X. L. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 5, 4477 (2014).
Ma, T. Y., Dai, S., Jaroniec, M. & Qiao, S. Z. Metal-organic framework-derived hybrid Co3O4-carbon porous nanowire arrays as reversible oxygen evolution electrodes. J. Am. Chem. Soc. 136, 13925–13931 (2014).
Hu, H., Guan, B. Y., Xia, B. Y. & Lou, X. W. Designed formation of Co3O4/NiCo2O4 double-shelled nanocages with enhanced pseudocapacitive and electrocatalytic properties. J. Am. Chem. Soc. 137, 5590–5595 (2015).
Deng, X. H. & Tüysüz, H. R. Cobalt-oxide-based materials as water oxidation catalyst: recent progress and challenges. ACS Catal. 4, 3701–3714 (2014).
Suntivich, J., May, K. J., Gasteiger, H. A., Goodenough, J. B. & Shao-Horn, Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science 334, 1383–1385 (2011).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Kanan, M. W. et al. Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy. J. Am. Chem. Soc. 132, 13692–13701 (2010).
Su, C. et al. SrCo0.9Ti0.1O3−δ as a new electrocatalyst for the oxygen evolution reaction in alkaline electrolyte with stable performance. ACS Appl. Mater. Interfaces 7, 17663–17670 (2015).
Risch, M. et al. Structural changes of cobalt-based perovskites upon water oxidation investigated by EXAFS. J. Phys. Chem. C 117, 8626–8635 (2013).
Zhu, Y. et al. SrNb0.1Co0.7Fe0.2O3−δ perovskite as a next-generation electrocatalyst for oxygen evolution in alkaline solution. Angew. Chem. Int. Ed. 54, 3897–3901 (2015).
Raccah, P. M. & Goodenough, J. B. First-order localized-electroncollective-electron transition in LaCoO3 . Phys. Rev. 155, 932 (1967).
Señarís-Rodríguez, M. A. & Goodenough, J. B. LaCoO3 revisited. J. Solid State Chem. 116, 224–231 (1995).
Korotin, M. A. et al. Intermediate-spin state and properties of LaCoO3 . Phys. Rev. B 54, 5309 (1996).
Zobel, C. et al. Evidence for a low-spin to intermediate-spin state transition in LaCoO3 . Phys. Rev. B 66, 020402 (2002).
Phelan, D. et al. Nanomagnetic droplets and implications to orbital ordering in La1−xSrxCoO3 . Phys. Rev. Lett. 96, 027201 (2006).
Hoch, M. J. R. et al. Diamagnetic to paramagnetic transition in LaCoO3 . Phys. Rev. B 79, 214421 (2009).
Křápek, V. et al. Spin state transition and covalent bonding in LaCoO3 . Phys. Rev. B 86, 195104 (2012).
Karolak, M. et al. Correlation-driven charge and spin fluctuations in LaCoO3 . Phys. Rev. Lett. 115, 046401 (2015).
Zhou, S. M. et al. Size-dependent structural and magnetic properties of LaCoO3 nanoparticles. J. Phys. Chem. C 113, 13522–13526 (2009).
Zhou, S. M. et al. Ferromagnetism in LaCoO3 nanoparticles. Phys. Rev. B 76, 172407 (2007).
Rossell, M. D. et al. Direct evidence of surface reduction in monoclinic BiVO4 . Chem. Mater. 27, 3593–3600 (2015).
Han, B. H. et al. Role of LiCoO2 surface terminations in oxygen reduction and evolution kinetics. J. Phys. Chem. Lett. 6, 1357–1362 (2015).
Gazquez, J. et al. Atomic-resolution imaging of spin-state superlattices in nanopockets within cobaltite thin films. Nano Lett. 11, 973–976 (2011).
Kwon, J. H. et al. Nanoscale spin-state ordering in LaCoO3 epitaxial thin films. Chem. Mater. 26, 2496–2501 (2014).
Lan, Q. Q. et al. Correlation between magnetism and ‘dark stripes’ in strained La1−xSrxCoO3 epitaxial films (0 ≤ x ≤ 0.1). Appl. Phys. Lett. 107, 242404 (2015).
Klie, R. F. et al. Direct measurement of the low-temperature spin-state transition in LaCoO3 . Phys. Rev. Lett. 99, 047203 (2007).
Qian, D. et al. Electronic spin transition in nanosize stoichiometric lithium cobalt oxide. J. Am. Chem. Soc. 134, 6096–6099 (2012).
Zhou, S. M. et al. Magnetic phase diagram of nanosized half-doped manganites: role of size reduction. Daton Trans. 41, 7109–7114 (2012).
Curiale, J. et al. Magnetic dead layer in ferromagnetic manganite nanoparticles. Appl. Phys. Lett. 95, 043106 (2009).
Wang, Y. & Fan, H. J. Low-field magnetoresistance effect in core–shell structured La0.7Sr0.3CoO3 nanoparticles. Small 8, 1060–1065 (2012).
Vasseur, S. et al. Lanthanum manganese perovskite nanoparticles as possible in vivo mediators for magnetic hyperthermia. J. Magn. Magn. Mater. 302, 315–320 (2006).
Wang, H. Y. et al. In operando identification of geometrical-site-dependent water oxidation activity of spinel Co3O4 . J. Am. Chem. Soc. 138, 36–39 (2016).
Malkhandi, S. et al. Design insights for tuning the electrocatalytic activity of perovskite oxides for the oxygen evolution reaction. J. Phys. Chem. C 119, 8004–8013 (2015).
Bao, J. et al. Ultrathin spinel-structured nanosheets rich in oxygen deficiencies for enhanced electrocatalytic water oxidation. Angew. Chem. Int. Ed. 127, 7507–7512 (2015).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Acknowledgements
This project was financially supported by the National Basic Research Programs of China (2012CB927402 and 2014CB932700), the National Science Foundation of China (Grant Nos. U1432134, 21203099, 51371164, and 21573206), the Anhui Provincial Natural Science Foundation (Grant No. 1508085QE109), the Collaborative Innovation Center of Suzhou Nano Science and Technology, Strategic Priority Research Program B of the CAS under Grant No. XDB01020000, Hefei Science Center CAS (2015HSC-UP016), Fundamental Research Funds for the Central Universities, the Doctoral Fund of Ministry of Education of China (20120031120033), the Research Program for Advanced and Applied Technology of Tianjin (13JCYBJC36800), and the Open Research Fund of State Key Laboratory of Luminescent Materials and Devices (2014-skllmd-05). We appreciate the support from the Tianjin Supercomputing Center.
Author information
Authors and Affiliations
Contributions
S.Z., Z.H. and J.Ze. designed the studies and wrote the paper. X.M., X.Z., C.M. and J.Zh. performed most of the experiments. Y.Q. and Z.H. carried out DFT calculations. S.Z., C.M., Z.H., L.S. and J.Ze. performed data analysis. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-12, Supplementary Tables 1-3, Supplementary Notes 1-3 and Supplementary References (PDF 2179 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhou, S., Miao, X., Zhao, X. et al. Engineering electrocatalytic activity in nanosized perovskite cobaltite through surface spin-state transition. Nat Commun 7, 11510 (2016). https://doi.org/10.1038/ncomms11510
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms11510
This article is cited by
-
High-spin Co3+ in cobalt oxyhydroxide for efficient water oxidation
Nature Communications (2024)
-
Understanding the sulphur-oxygen exchange process of metal sulphides prior to oxygen evolution reaction
Nature Communications (2023)
-
Effect of Electron Structure of La-Based Perovskites on the Catalytic Combustion of n-Butylamine
Catalysis Letters (2023)
-
Electronic Structure and Magnetocaloric Effect of Sr-Doped SmCoO3 Perovskites
Journal of Electronic Materials (2023)
-
La0.75Sr0.25MnO3-based perovskite oxides as efficient and durable bifunctional oxygen electrocatalysts in rechargeable Zn-air batteries
Science China Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
