
Abstract
Proteins of the bromodomain and extraterminal (BET) family, in particular bromodomain-containing protein 4 (BRD4), are of great interest as biological targets. BET proteins contain two separate bromodomains, and existing inhibitors bind to them monovalently. Here we describe the discovery and characterization of probe compound biBET, capable of engaging both bromodomains simultaneously in a bivalent, in cis binding mode. The evidence provided here was obtained in a variety of biophysical and cellular experiments. The bivalent binding results in very high cellular potency for BRD4 binding and pharmacological responses such as disruption of BRD4–mediator complex subunit 1 foci with an EC50 of 100 pM. These compounds will be of considerable utility as BET/BRD4 chemical probes. This work illustrates a novel concept in ligand design—simultaneous targeting of two separate domains with a drug-like small molecule—providing precedent for a potentially more effective paradigm for developing ligands for other multi-domain proteins.
Similar content being viewed by others
Main
The bromodomain (BRD)-containing proteins are a family of proteins responsible for reading epigenetic acetylated lysine marks on histones and are rapidly emerging as a target class amenable to inhibition by small molecules1. The proteins in the BET subfamily are perhaps the most widely explored of the class2. Target identification and optimization of compounds initially identified as a result of phenotypic screening for modulation of apolipoprotein A1 have revealed multiple small-molecule inhibitors of BET BRDs which have progressed to preclinical and clinical evaluation3,4,5.
The BET family consists of four proteins, termed BRD2, BRD3, BRD4 and BRDT6. Each of these proteins contains two separate BRDs7. Existing chemical probes, such as i-BET762 (1) (Fig. 1a), do not show selectivity for individual family members and are monovalent ligands, which bind monovalently to both BRDs within each protein with a 2:1 stoichiometry. These chemical probes have been critical for elucidating underlying BET biology. In oncology, the existence of tumor types driven by BRD4 fusion mutations5 and those in which critical oncogenes such as c-Myc are regulated by BRD4 (refs. 8,9) has led to great interest in BRD4/BET inhibitors, and a number are currently progressing through early clinical trials10.

(a) Prototypic BET inhibitor i-BET762 (1) and potent AR downregulator 2. DR, downregulation. (b) BRD selectivity data for 2 showing the binding percentage at 10 μM; numbers above bars denote pKd values. (c) Concentration response for c-Myc downregulation determined by western blotting for 1 and 2 in MM.1S cells (additional details are provided in the Online Methods).
Here we describe our discovery of a new class of bivalent BET inhibitors, culminating in the chemical probe biBET (6). Using a combination of techniques, we determined that they act via an in cis binding mode, simultaneously engaging both BRDs in a single protein. This binding mode leads to enhanced cellular potency and slow dissociation kinetics, resulting in what to our knowledge is the most potent cellular probe for BET proteins described so far.
Results
Discovery of bivalent BET inhibitors
As part of a program to identify downregulators of the androgen receptor (AR), the development candidate AZD3514 was recently reported11,12. AZD3514 displayed binding to the AR and cellular AR downregulation at similar and reasonably high concentrations. We therefore embarked on a chemistry campaign focused on increasing potency, leading to the discovery of compounds such as 2, which demonstrated increased AR downregulation potency but showed a disconnect between binding and cellular potency values (Fig. 1a). Moreover, it was revealed that these compounds also effected downregulation of estrogen receptor-α (ERα) at concentrations comparable to those seen for the AR, suggesting that there was indirect and unknown pharmacology driving both of these phenotypes.
These findings led us to consider what the direct target of the compounds might be. Comparison of these structures with known BET protein inhibitors led us to speculate that elements of our compounds might be capable of binding BRDs and that this might have been the cause of the observed pharmacology. For instance, the chlorotriazole motif contained within the triazolopyridazine (TPDZ) of 2 could be overlayed with the methyltriazole of 1. Alternatively, the piperazinone N-methyl and carbonyl groups could be overlayed with those of acetyl-lysine. Compound 2 was subsequently tested in a panel of BRD assays13, which revealed activity against BET-family proteins (pKd values of 7.2 against BRD4 isolated bromodomain 1 (BRD4(1)) and 6.1 against BRD4 isolated bromodomain 2 (BRD4(2))) (Fig. 1b). Further profiling in cells showed that 2 caused downregulation of c-Myc (Fig. 1c) and inhibition of tumor cell growth in a manner consistent with sensitivity to BRD4 inhibition, that is, complete inhibition of cell growth and strong induction of apoptosis in BRD4-sensitive MM.1S, MOLP-8, MV-4-11 and RS4;11 cells (pGI50 (−log of the concentration that causes 50% net growth inhibition) values of 7.3, 7.2, 7.4 and 6.8, respectively); partial growth inhibition without discernible apoptosis induction was observed in less sensitive K-562 cells (pGI50 = 6.0).
We found that 2 also had weak activity for BRD4(1) and BRD4(2), but with values comparable to its AR-binding and downregulation potencies. It is interesting to note that an independent analysis subsequently revealed that 2 has a pharmacological profile similar to those of BET inhibitors in cancer cell panels14,15. The ability of BET inhibitors to effect inhibition of AR-mediated signaling16 and ERα downregulation17 has since been reported.
Induced dimerization of BRD4(1) by X-ray crystallography
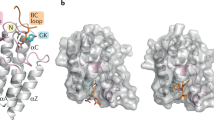
Our structural hypotheses implied that either the TPDZ or the piperazinone group could engage the BET acetyl-lysine pocket. Solving the co-crystal structure of 2 in the presence of BRD4(1) (PDB ID 5AD2; Supplementary Results, Supplementary Table 1 and Supplementary Fig. 1) revealed a remarkable compound-induced dimerization of two BRD4(1) molecules within the crystal lattice, with 2 spanning both acetyl-lysine binding sites (Fig. 2a). The TPDZ group was observed to make hydrogen bonds with the side chain of Asn140 and a structural water molecule at the base of the pocket (Fig. 2b). The piperidine ring projected over the 'WPF shelf' (W81, P82, F83)3, and the aryl ring contacted the WPF shelf of the second BRD with the pendant piperazinone engaging the second binding site. The piperazinone formed a strong H-bond with a structural water molecule and a weaker polar contact with the side chain of Asn140 (Fig. 2c).

(a) The compound induced dimerization. (b) Interactions made by the TPDZ group in the acetyl-lysine pocket. (c) Interactions made by the piperazinone in the second acetyl-lysine pocket. Hydrogen bond distances (green dashed lines) are shown.
Optimization of bivalent binding
The dimeric X-ray structure was consistent with the structure–activity relationships (SARs) we had observed in cellular assays, with changes to both ends of the molecule that removed the observed interactions with the acetyl-lysine pocket resulting in a loss of potency. For example, a compound in which the TPDZ chloro-substituent (postulated to mimic the methyl of acetyl-lysine) was replaced with an alternative methyl mimic, such as a trifluoromethyl group (3), retained activity, whereas deletion of the substituent (4) led to a loss of BRD4 affinity and cellular potency (Fig. 3a). Moving the carbonyl substituent of the piperazinone to the 3 position (5), thereby disrupting the interaction with the critical Asn residue, led to a loss of cellular potency but with retained BRD4(1)-binding affinity (details of compound synthesis are presented in Supplementary Note 1).

(a) Deletions of BRD binding elements. DR, downregulation; ND, no data. (b) Optimized bis-TPDZs 6 and 7 and inactive control compound 8 modified to abrogate both binding events. (c) Concentration response overlays for 6 in Bromoscan competition binding assays for BRD4(1) (circles), BRD4(2) (squares) and BRD4(1,2) (triangles). (d) X-ray structure of BRD4(1) dimer induced by 6 (PDB ID 5AD3) (Supplementary Table 4).
We reasoned that, in a bivalent complex, one of the BRD-binding motifs may possess higher affinity than the other and pseudosymmetrical compounds may have further increased potency. The data for binding to BRD4(1) for 4 and 5 suggested that the TPDZ was the more potent warhead, given the greater loss of potency in 4 compared with 5. Accordingly, we designed and synthesized compounds with a second TPDZ motif replacing the piperazinone of 3, resulting in pseudosymmetrical compounds 6 and 7 (Fig. 3b). In this case, a methoxy-substituted TPDZ was used, as we found that this substituent led to improved physical properties without a loss of potency. These compounds showed increased potency against BRD4(1), along with further increased potency against the tandem domain construct (pKd = 11; Fig. 3c) and in cells (e.g., pEC50 values of 9.2 and >9.3 for c-Myc downregulation for 6). The compounds also retained a BRD-selectivity profile similar to that of 3 (Supplementary Fig. 2). These cellular potencies are, to the best of our knowledge, the highest reported to date for BRD4 inhibitors, with 6 and 7 more than two orders of magnitude more potent than 1. Rearrangement of the acetyl-lysine mimic of the triazolopyridazine coupled with inversion of the stereocenter (8) led to considerably reduced BRD4 activity and no detectable ERα downregulation, providing an inactive control and further evidence that the observed pharmacology was BRD4 driven. Compound 7 proved too insoluble (<1 μM) for further biophysical characterization, and so in further studies we focused on 6, sebseqently termed biBET, which showed good in vitro ADME (absorption, distribution, metabolism and excretion) properties (Supplementary Table 2).
In selectivity panels of 32 BRDs13, 123 kinases and 147 secondary pharmacology assays, 6 showed selectivity for the BET family compared with other BRDs (Supplementary Fig. 2) and no detectable kinase activity at 1 μM (Supplementary Table 3). Only ten of the other secondary pharmacology assays registered potencies below 10 μM (Supplementary Table 4), with the most potent being the 5HT1B receptor (agonist effector concentration for half-maximum response (EC50) of 0.93 μM; selectivity ratio of 1,400-fold relative to BRD4 potency).
The X-ray structure of 6 with BRD4(1) revealed a dimeric structure similar to that observed with 2 (PDB ID 5AD3; Fig. 3d, Supplementary Table 5 and Supplementary Fig. 3). The triazolopyridazine groups were seen to engage in differential interactions in the two BRDs. One group made similar contacts with Asn140 and the structural waters as observed with 2, whereas in the other binding site the hydrogen bonds to the waters were observed but that to Asn140 was not (Supplementary Fig. 4). The latter is not what is typically seen with triazolopyridazine structures and might not represent the mode of engagement that occurs in solution. Indeed, a close analog of 6 was observed to engage with Asn140 and the key structural water with both TPDZs under the same conditions (Supplementary Fig. 5).
Determination of compound binding mode
The enhanced potency of 6 and 7 against the tandem domain constructs and in cellular assays led us to believe that the bivalent binding observed in the X-ray structure was relevant to their cellular mechanism of action. Given the presence of two BRDs in full-length BRD4, there are several possible binding modes for such bivalent compounds. In one mode, the compounds may bind with 2:1 stoichiometry with one compound occupying each BRD in the same way as observed for monovalent compounds such as 1 (Supplementary Fig. 6a). This was considered unlikely to be the sole binding mode, as it would not explain the observed SARs or the difference in behavior relative to monovalent ligands such as 1. Alternatively, the two warheads of a single molecule may bind to a BRD to induce an in cis complex (Supplementary Fig. 6b) or an in trans dimer (Supplementary Fig. 6c). In the case of in trans binding, multiple possibilities exist with different combinations of each warhead binding to BD1 and BD2. In addition, higher-order multimeric species may form.
A computational model of the full-length protein based on the structure of 2 using a homology model of BD2 and a simulated interdomain region showed that formation of an in cis complex (Supplementary Fig. 6d) as well as an in trans dimer (Supplementary Fig. 6e) was possible (Online Methods). Under physiological conditions, any one or more of these states may exist, and the relative population of each is likely to vary with both protein and compound concentration. In particular, the 2:1 complex is likely to dominate at very high concentrations as the protein becomes saturated with ligand.
Binding of 1 and 6 to BRD4(1,2) tandem domain by NMR
The first step in elucidating the binding mode of the compounds was to establish whether both BRDs were engaged by the ligands in solution. We recorded a series of two-dimensional transverse relaxation optimized 1H–15N heteronuclear single quantum coherence (TROSY HSQC) NMR spectra on tandem domain BRD4(1,2) (residues 44–460) in complex with monovalent 1 and bivalent 6 at ligand-to-protein ratios of 1:1 and 2:1 (Fig. 4a). BRD4(1,2) showed a chemical shift dispersion characteristic of a structured protein with an unstructured region (Supplementary Fig. 7a). We were able to assign tryptophan resonances by comparison of the individual spectra for the isolated first and second BRDs (BD1 and BD2, respectively) with that of the tandem domain (Supplementary Fig. 7b), allowing the W374 resonance in BD2 to be assigned and the remaining three tryptophan resonances to be ascribed to BD1 (Online Methods). We obtained independent confirmation of resonances from BRD4(2) through segmental isotope labeling of the C-terminal BRD of BRD4(1,2)18.

(a) 2D 1H–15N TROSY HSQC spectra recorded at 18.8 T of U-[2H,15N]-labeled BRD4(44–460) (gray) compared to the spectra in the presence of 1 (red, left) and 6 (blue, right) at a ligand:protein ratio of 2:1. Boxed regions are shown expanded in Supplementary Figure 7c and correspond to (i) a BD1 residue (unassigned) and (ii) the W374 indole amine of BD2. (b) Sedimentation coefficient distributions for apo-BRD4(1–477) (gray) and in the presence of 1 and 6 at 1:1 and 2:1 ligand:protein ratios. (c) Sedimentation coefficient distributions for 6 at a 1:1 ligand:protein ratio overlaid with the contour heat map of likely frictional ratios across the peaks. (d) Ab initio shapes of the BRD4 constructs restored from the SAXS data for apo-BRD4 and 1:1 complexes with 1 and 6; distances are presented as mean ± s.d. (n = 100). Additional data on interdomain distances, Rg values derived from model fitting and χ2 values showing the goodness of fit are given in Supplementary Table 6.
Shifts in the signals for both domains were seen clearly in the presence of 1 in the slow-exchange regime typical for tight binding (Fig. 4a and Supplementary Fig. 7c). Similarly, upon titration of 6, engagement of both domains simultaneously was observed. Given the difference between the affinities of 6 measured for BRD4(1) and BRD4(2), this observation is inconsistent with monovalent binding, with which a greater proportion of engagement with BD1 would be expected. In addition, binding of 6 markedly changed a number of other peaks in the spectrum, which were very different from those observed for 1. Together, these observations show that 6 binds to both BD1 and BD2 acetyl-lysine pockets and suggest that its binding mode is different from that of 1 (Supplementary Note 2).
Characterization of complexes of 1 and 6 by AUC and SAXS
Analytical ultracentrifugation (AUC)19 for apo-BRD4(1,2) showed a sedimentation coefficient (s20,W) of 2.46 ± 0.05 S and an f/f0 ratio of 2.2, consistent with a dispersed monomer of highly extended conformation. In the presence of 1, the sedimentation coefficient was very similar to that of apo-BRD4(1,2), indicating no change in oligomeric state or gross conformation (Fig. 4b). In the presence of 6, however, dramatic effects on the peak profiles and sedimentation coefficients were observed. At 1:1, two peaks were observed: one with a sedimentation coefficient similar to that of apo-BRD4(1,2), and a faster-sedimenting species (s20,W = 2.8 S). To test whether these changes were due to conformational changes in the molecule, we analyzed the data using a c(s, f/f0) model20. The contour plot of the distribution of frictional ratios across the peaks of the 1:1 complex with 6 (Fig. 4c) showed that the frictional ratio of the species within the two peaks varied from ∼2.2 for the first peak to ∼1.4 for the second, indicative of conformational tightening of the molecule. At a 2:1 ligand:protein ratio, the same analysis revealed the formation of smaller populations of dimeric species (Supplementary Fig. 8). These sedimentation analyses indicated that binding of 6 elicits a large conformational change in BRD4, resulting in a more compact shape, and also suggested the formation of a small proportion of dimeric species at higher ligand concentrations. These species probably represent in cis and in trans interactions between the BRDs induced directly by 6.
Small-angle X-ray scattering (SAXS)21 studies on apo-BRD4(1,2) showed it to be largely monomeric at concentrations below 1 mg/ml (at higher concentrations, the protein tended to aggregate in both the presence and the absence of compound, and so the data were extrapolated to zero concentration for analysis) and flexible in solution. Ab initio and ensemble optimization method analysis22 suggested that it was extended (Fig. 4d). This changed very little after the addition of 1. In the presence of 6, the protein became progressively and noticeably more compact. The most compact complex was obtained at a 2:1 ligand:protein ratio, indicating that the greatest population of in cis complex was present at this stoichiometry.
We calculated distributions of interdomain distances (between Ile110 in BD1 and Met425 in BD2) from the 100 out of 10,000 theoretical models best fitting the experimental data (Online Methods). In the apo protein, we observed an average interdomain distance of 140 ± 12 Å (Fig. 4d), representing an absence of contacts between the two domains, and the complex with 1 showed only slightly smaller distances. In contrast, the complex with 6 had significantly shorter interdomain separations (average of 84 ± 18 Å and 63 ± 18 Å for the 1:1 and 2:1 complexes, respectively; P ≤ 0.00001). The radii of gyration (Rg) showed a similar picture (Supplementary Table 6). These data are strongly suggestive of a different binding mode for 6 compared to 1, and one in which a substantial change in protein conformation is induced.
The comparable interdomain distance in the BD1 dimeric crystal structure of 6 was 38 Å. This is consistent with the formation of a complex with BD1 and BD2 in similar proximity in the SAXS experiment. As the Rg values for 6 were lower than those for the apo protein and complexes with 1, it is reasonable to assume that the complex with 6 was mostly monomeric. Taken together, these findings are highly suggestive of the formation of a substantial population of an in cis complex. However, it must be noted that these data have been extrapolated to infinite dilution and therefore might not reflect the binding mode in cellular conditions, under which higher-order multimers may form.
Cellular mode of action and pharmacology
We developed bioluminescence resonance energy transfer (BRET) systems23 in HCT116 and HEK293 cells to monitor interaction with chromatin using Halo-tagged histone H3 and a nanoluciferase fusion protein with either BRD4(1) or full-length BRD4. Compound 1 displaced both isolated BRD4(1) and the full-length protein with similar potency (Fig. 5a). In contrast, 6 showed a remarkable (four orders of magnitude) enhancement in potency for displacement of full-length BRD4 relative to BRD4(1) (pIC50 (−log of the half-maximal inhibitory concentration) of 9.6 and 5.6, respectively). This potency increase provides evidence that the bivalent mechanism also operates in cells and is responsible for the cellular potency of 6. Analogous experiments comparing full-length wild-type BRD4 to the N433F point mutant (BRD4(N433F)) provided compelling evidence of the need to engage BD2 specifically to exert this effect (Fig. 5a). The mutant showed a chromatin-displacement response similar to that of isolated BRD4(1) in these experiments, and the potency of 6 was similar to that of 1 (pIC50 values of 6.6 and 6.8, respectively). The binding of BRD4 to acetylated chromatin is believed to be dominated by BD1 interactions24, and the N433F mutation is known to provide a steric barrier to engagement of the BRD4(2) acetyl-lysine pocket25, so bivalent engagement of both acetyl-lysine pockets by 6 is required to exert the increased potency.

(a) NanoBRET concentration response curves for displacement of BRD4(1), full-length (FL) BRD4 and BRD4(N433F) from labeled histone H3.3–HaloTag by 1 and 6 comparing BRD4(1) to full-length BRD4 in HCT116 cells (left) and full-length BRD4(1) to BRD4(N433F) in HEK293 cells (right). (b) NanoBRET sensor containing BRD4(1,2) with HaloTag (HT) BRD4(1) and nanoluciferase (NL) BRD4(2) showing increased BRET response for wild-type BRD4(1,2) in the presence of 6, which was not observed for the N140F mutant or for binding of 1. (c) Intracellular NanoBRET target engagement using a fluorescent iBET-762-based tracer. The left graph shows competitive displacement from full-length BRD4, BRD4(N433F) and BRD4 (1) in HeLa cells by 1 and 6. The other three graphs present results of residence time analysis for compounds 1 and 6 on BRD4(1), full-length BRD4 and BRD4(N433F) in HeLa cells. Data in all graphs are the mean and s.e.m. of three independent determinations.
To detect whether binding of 6 produced a conformational change in BRD4, we constructed a NanoBRET BRD4 biosensor consisting of the two tandem BRDs, BD1 and BD2, expressed between HaloTag and NanoLuc fusions (Fig. 5b). The signal in this conformational biosensor showed no change after treatment with 1 compared to basal levels. Treatment with 6, however, led to a considerable increase in signal at the concentration causing chromatin displacement. The plateau in signal observed with 6 was highly suggestive of an in cis binding mechanism. To study this further, we made a point mutant within BD1, N140F, within the biosensor. This mutant biosensor demonstrated a basal-level signal similar to that of the wild type, no change in signal after treatment with 1 (Fig. 5b) and, strikingly, no response to 6. Together these data support in cis binding of 6 to both BRDs within a single BRD4 protein.
To analyze intracellular target engagement of the compound with BRD4, we monitored BRET using a fluorescently labeled version of 1 in conjunction with nanoluciferase fusion constructs of BRD4(1), full-length BRD4 and BRD4(N433F) competitively displaced with varying concentrations of test compound26 (Fig. 5c). Again, 6 showed a remarkable difference in potency for displacement of tracer from full-length BRD4 relative to BRD4(1) and BRD4(N433F) (pIC50 values of 9.0, 5.8 and 6.3, respectively). The same was not observed for 1. As anticipated, comparative experiments assessing intracellular target engagement (Supplementary Fig. 9) and acetylated histone displacement (Supplementary Fig. 10) for the other BET-family proteins showed similar effects.
We then determined the intracellular residence times of 1 and 6. For this we incubated cells expressing nanoluciferase fusions of BRD4 with the appropriate single concentration, equivalent to the corresponding IC50, of compound for 3 h (1 nM 6 used for full-length BRD4 and 1 μM 6 used for BRD4(N433F) and BRD4(1)). After compound equilibration and multiple wash steps, we added 1 μM tracer to intact cells and measured the dissociation of 1 and 6 (ref. 26). Compounds 1 and 6 showed rapid dissociation from BRD4(1) and BRD4(N433F), but 6 showed slower displacement from full-length BRD4 (Fig. 5c). Thus the bivalent binding also led to an extended residence time compared with monovalent ligands.
We next studied the disruption of BRD4 foci in native U2OS cells. We observed bright BRD4 foci with immunostaining using an antibody to BRD4 (Fig. 6a) and confirmed the specificity of the antibody via short interfering RNA knockdown of BRD4. Recent literature has described asymmetrical loading of BRD4 to super-enhancers, which are often associated with key genes that determine cell fate or are involved in oncogenesis27,28. Histone H3K27 acetylation is a marker for active enhancers29. Mediator complex subunit 1 (MED1) and BRD4 have previously been shown to demonstrate high binding to super-enhancers, and their levels are strongly correlated throughout the genome30. Consistent with this, immuno-costaining experiments showed that MED1 and H3K27Ac formed foci in U2OS cells that overlapped completely with those of BRD4.

(a) Formation of bright foci from super-loading of BRD4 or MED1 to the super-enhancers marked by histone H3K27Ac in the chromatin regions. (b) Representative images showing disruption of BRD4 and MED1 foci by 6 (10 nM). (c) Growth inhibition data for 1 and 6 in BRD4-sensitive (MM.1S; left) and BRD4-insensitive (RS4;11; right) cell lines. Error bars represent the s.d. of triplicate determinations. Full curves for the other tested lines are shown in Supplementary Figure 12. (d) c-Myc protein levels measured by western blotting for 1 (1 μM) and 6 (50 nM) compared to DMSO control at 2, 4 and 16 h after compound washout in MM.1S cells (full-length gels are shown in Supplementary Fig. 13).
Treatment with 10 nM 6 led to complete disruption of BRD4 and MED1 foci (Fig. 6b). Concentration-response data for 1 and 6 in this assay showed that 6 was a far more potent inhibitor (pIC50 values of 7.4 and 10 for 1 and 6, respectively; Supplementary Fig. 11). There was no apparent change of H3K27Ac foci with up to 24 h of treatment with BRD4 inhibitors, whereas the disruption of BRD4 and MED1 foci reached maxima within 3–4 h.
The enhanced potency of 6 in BRD4-related assays translated into cancer-cell-killing activity in a mixed panel of hematological cell lines (Fig. 6c and Supplementary Fig. 12). The bivalent binding of 6 translated into highly potent cell-killing activity in BRD4-dependent cell lines (pIC50 of 9.5 in both MV4.11 and MM.1S cells), representing a potency increase of approximately three orders of magnitude compared to 1. Activity in BRD4-independent K-562 cells was weaker than in BRD4-sensitive lines with both 1 and 6. Interestingly, the acute lymphoblastic leukemia cell line RS4-11 was quite sensitive to both BET inhibitors. However, monovalent 1 did not achieve full inhibition, even at higher concentrations, whereas bivalent 6 achieved near-complete cell killing. This provides evidence of differentiated and possibly advantageous pharmacology from bivalent inhibition. Changes in gene expression in response to treatment with 1 and 6 in MM.1S, MV-4-11 and K-562 cells revealed very similar expression profiles in response to both inhibitors (Online Methods), suggesting that the enhanced cell-killing effects of 6 are a direct consequence of the increased inhibitory potency as a result of bivalent inhibition rather than altered downstream gene expression.
To further corroborate the extended intracellular BRD4 residence time observed for 6, we performed a drug washout using MM.1S cells and monitored c-Myc protein recovery over time (Fig. 6d). This clearly showed that 6 maintained knockdown of c-Myc protein levels up to 16 h after washout. In contrast, after treatment with 1, c-Myc levels returned to control levels within 1 h of washout.
Discussion
This study revealed unique BET inhibitors (biBETs) that simultaneously engage two BRDs, leading to biochemical and cellular consequences beyond those shown with monovalent inhibitors. Enhanced cell potency correlated across a range of cell formats and end points including AR and ERα immunofluorescence in prostate and breast cancer cells, respectively, transiently expressed engineered BRET protein–protein interaction systems and well-characterized BRD4-related end points such as c-Myc downregulation in leukemia cells (Supplementary Table 7). The potency and selectivity achieved by biBET 6, coupled with the comprehensive evidence for its enagement with BRD4 in cells, make it the best cellular probe reported to date (to our knowledge) when judged according to recently described criteria for chemical probes31.
The results we have described provide compelling evidence for the bivalent mechanism. The cellular SAR for the analogs is highly suggestive of 6 binding two BRDs simultaneously. The X-ray crystal structures of the compounds in complex with an isolated BRD4(1) construct clearly demonstrate their ability to induce dimerization of two BRDs. The NMR experiments on tandem domain constructs showed that 6 engaged both BD1 and BD2 of BRD4 simultaneously in solution. The AUC data demonstrate that binding of 6 led to a protein with a smaller radius than either the apo protein or that in complex with a monovalent ligand, suggesting that the bound complex in solution is mostly monomeric with respect to protein and that compound binding induces a more compact structure. SAXS experiments showed folding of the protein in response to ligand binding and demonstrated that the complex with 6 brought the two BRDs into close proximity, similar to what was observed in the X-ray structures of the BRD4(1) dimer. Together, these data provide strong evidence for the formation of substantial populations of in cis complex under the conditions studied. The dramatic increase in potency observed in the cellular NanoBRET experiment for displacement of BRD4(1,2) from acetylated histone (relative to the BD1 construct alone), the analogous decrease in BRD4(1,2) binding potency with the BRD4(2) binding-site-blocking mutation (N433F) and subsequent translation of the increased potency into BRD4-driven pharmacology, along with an increased BRET response for binding to doubly tagged BRD4(1,2), provide very strong evidence that 6 binds bivalently in cells with a substantial population of an in cis complex forming.
Data on cellular growth inhibition showed that differential and potentially advantageous pharmacology arose from the bivalent binding mode: complete inhibition of less sensitive cell lines was observed with 6, whereas monovalent ligands did not achieve complete inhibition in the same settings. This could be a result of bivalent binding per se or as a follow-on consequence of the slow dissociation arising from the bivalent mechanism. We anticipate that these effects would translate to in vivo settings, although this would require optimization of pharmacokinetic profiles. The pharmacology of bivalent inhibitors with optimized in vivo exposure is the subject of other publications32,33.
The increased potency due to bivalent binding is presumably an avidity effect34. Past attempts to produce bivalent binders to BRDs35,36 and other proteins37 have involved linking two separate small-molecule ligands with a flexible linker. Compounds such as 6 that partially fuse the two binding elements result in much improved physical properties (6 has a molecular weight of 531 and logD7.4 of 2.9); thus these compounds could conceivably be optimized as drugs. Many proteins possess an arrangement of ordered functional domains joined by extended disordered regions. In particular, in the epigenetics field, protein–histone interactions achieve their affinity and specificity through the sum of weak interactions with different epigenetic marks38,39,40,41. The development of bivalent ligands for these proteins in a manner analogous to that described here could provide a means of developing superior chemical probes and, ultimately, drug molecules (Supplementary Fig. 14a) and could also provide a more effective means of targeting protein–protein complexes (Supplementary Fig. 14b).
Starting with an initial hit molecule with an unknown target, we were able to follow cellular SARs to elucidate the molecular mechanism. Further optimization based on our mechanistic hypothesis enabled us to substantially improve the compounds' effectiveness by structure-based design, increasing their potency from micromolar to picomolar as a consequence of bivalent target engagement. As a result, we have created biBET ligands that can bind simultaneously to two BRDs. It is also notable that these bivalent molecules deliver a very substantial increase in potency while maintaining acceptable size and physical properties. The resulting compound is a very potent, selective inhibitor of BET BRDs and provides a chemical probe with distinct advantages over previously described probes. We anticipate that this will be invaluable in further elucidating the complex biology of BET proteins.
Methods
In vitro ADME data for 6.
Data were generated using standard protocols42,43.
Protein preparation.
Plasmid constructs.
Variants of human BRD4 were generated by gene synthesis with an N-terminal hexahistidine tag and tobacco etch virus (TEV) protease site and subcloned into pET28 vector for Escherichia coli expression. Variants included the following: BRD4(1,2), residues 1–477; BRD4(1,2), residues 44–460; BRD4(1), residues 42–169; and BRD4(2), residues 333–460.
Protein expression.
In E. coli cells (BL21-Gold (DE3); Novagen), BRD4 proteins were induced using 0.1 mM IPTG overnight at 18 °C.
Protein purification.
Cells were resuspended in 50 mM Tris, pH 8.0, 300 mM NaCl, 1 mM β-mercaptoethanol, 10 mM imidazole containing Complete Protease Inhibitor tablets (Roche) and benzonase nuclease (2.5 u/ml).Resuspended cells were lysed using a Constant Systems cell disruptor at 25 k.s.i. and clarified by centrifugation at 75,000g for 2 h at 4 °C.
BRD4 was purified from the supernatant by nickel-affinity chromatography followed by cation-exchange chromatography. Protein was further purified by size-exclusion chromatography using a Superdex 75 10/300 GL column in 20 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM Tris(2-carboxyethyl) phosphine hydrochloride.
For NMR spectroscopy and X-ray crystallography, the hexahistidine tag was removed from BRD4 by treatment with TEV protease after the nickel-affinity chromatography.
Crystallization and structure of BRD4(1) in complex with 2.
E. coli–expressed BRD4(42–169) at a concentration of 10 mg/ml was mixed with 2 to a final concentration of 200 μM, 2% DMSO and incubated on ice for 1 h. Note that owing to limited solubility and DMSO tolerance, a molar excess of the compound concentration could not be attained. We carried out low-volume sitting-drop crystallization in MRC two-well plates using a ratio of 2:1 protein:mother liquor, and crystals formed in a condition containing 0.2 M NaCl, 5.9% PEG 3350 (wt/vol), 10% ethylene glycol, 0.1 M HEPES, pH 7.4, at 4 °C. Crystals were cryoprotected in mother liquor supplemented with an additional 20% ethylene glycol. Diffraction data were collected using a Rigaku FRE superbright generator with Osmic mirrors and a Saturn 944 CCD (charge-coupled device) detector and processed using XDS44. Molecular replacement solution was determined with 2OSS.pdb using Phaser45. The P212121 cell (α = β = γ = 90°, a = 41.618 Å, b = 59.615 Å, c = 106.773 Å) has a dimer in the ASU. Model building was performed using Coot46 and refined using Buster47. Compound 2 could easily be modeled into the difference density using ligand restraints generated by Grade48. It should be noted that the electron density for the ligand was of sufficient quality to enable it to be built in two alternate and equivalent conformations with a total final occupancy of 1.0. The statistics for the data collection and refined coordinates are given in Supplementary Table 1. The final model has been deposited in the Protein Data Bank under accession code 5AD2.
Crystallization and structure of BRD4(1) in complex with 6.
E. coli–expressed BRD4(42–169) at a concentration of 10 mg/ml was mixed with 6 to a final concentration of 200 μM, 2% DMSO and incubated on ice for 1 h. Note that owing to limited solubility and DMSO tolerance, a molar excess of the compound concentration could not be attained. We carried out low-volume sitting-drop crystallization in MRC two-well plates using a ratio of 2:1 protein:mother liquor, and crystals formed in a condition containing 0.2 M (NH4)2HPO4, 20% PEG 3350 (wt/vol) at 4 °C. Crystals were cryoprotected in mother liquor supplemented with an additional 20% ethylene glycol. Diffraction data were collected at beamline IMCA-CAT 17ID at the Advanced Photon Source at Argonne National Laboratory (Argonne, Illinois, USA) using a Dectris Pilatus 6M pixel array detector and were processed using XDS. Molecular replacement solution was determined with 2OSS.pdb using Phaser. The P212121 cell (α = β = γ = 90°, a = 109.64 Å, b = 41.826 Å, c = 59.591 Å) has a dimer in the ASU. Model building was performed using Coot and refined using Buster. Compound 6 could easily be modeled into the difference density bound in a single orientation at occupancy of 1.0 using ligand restraints generated by Grade. The statistics for the data collection and refined coordinates are given in Supplementary Table 5. The final model has been deposited in the Protein Data Bank under accession code 5AD3.
Measurement of endogenous ERα levels in the MCF-7 cell line.
We measured endogenous ERα levels in the MCF-7 cell line as previously described49. Briefly, we revived the MCF-7 human breast ductal carcinoma cell line (American Type Culture Collection (ATCC); HTB-22) directly from a cryovial in phenol-red-free Dulbecco's modified Eagle's medium (DMEM) containing 2 mM L-glutamine and 5% (vol/vol) charcoal/dextran-treated fetal calf serum (FCS). Cells were seeded into black, transparent-bottomed, 384-well plates at 1,500 cells per well in a volume of 40 μl using a Multidrop Combi dispenser (Thermo Scientific). Cell plates were incubated for 24 h at 37 °C, 5% CO2. An Echo 555 acoustic dispenser was used to dispense test compounds directly into cell plates, producing a 12-point half-log duplicate concentration range (3.125 μM top concentration). DMSO was dispensed in the maximum-signal control wells to give a final concentration of 0.3% (vol/vol), and fulvestrant was dispensed in the minimum-signal control wells to give a final concentration of 100 nM. Cells were incubated for 18–24 h at 37 °C, 5% CO2 and then fixed using a 3.7% (vol/vol) final formaldehyde concentration for 20 min. The cells were washed three times with 250 μl of phosphate-buffered saline solution (PBS) using a BioTek ELx405 plate washer (BioTek Instruments, UK), and 50 μl of PBS was added to all wells. The PBS was aspirated and the cells were permeabilized with 40 μl of PBS containing 0.5% (vol/vol) Tween 20 for 1 h at room temperature. The cells were then washed three times in 250 μl of PBS with 0.05% (vol/vol) Tween 20 (PBST) using a BioTek EL406 plate washer. We added 20 μl per well of a 1:1,000 dilution of ERα monoclonal rabbit antibody (clone SP1 (epitope contained within a synthetic peptide derived from C-terminal domain of human estrogen receptor); Thermo Scientific, RM-9101-S) in PBST with 3% (wt/vol) bovine serum albumin (BSA) and incubated the plates overnight at 4 °C. The cells were washed three times in 250 μl of PBST and then incubated with 20 μl per well of a 1:500 dilution of goat anti-rabbit IgG conjugated to Alexa Fluor 594 (B40944; Molecular Probes, Life Technologies, Paisley, UK) and a 1:5,000 dilution of Hoechst (Molecular Probes) in PBST with 3% (wt/vol) BSA for 1 h at room temperature. The cells were washed three times in 250 μl of PBST; then 40 μl of PBS was added per well and the plates were covered with a black plate seal. Cell images were acquired using a Cellomics Arrayscan VTI HCS reader (Thermo Scientific) using a 10×/0.3-NA objective and applying the filters BGFR_386_23 and BGFR_549_15 to detect Hoechst and Alexa Fluor 594 signals. Using the Compartmental Analysis v4 bio-application, we generated nuclear masks from the Hoechst-stained nuclei in channel 1 and applied a suitable threshold in channel 2 to quantify ERα within the nuclear masks. The mean total intensity was normalized for cell number, giving the total intensity of ERα staining per cell.
Measurement of endogenous AR levels in the LNCaP cell line.
96-well method using monoclonal mouse anti-human AR.
We used a modification of the previous immunofluorescence end-point assay11 to measure levels of nuclear AR in the LNCaP prostate carcinoma cell line (clone FGC (CRL-1740); ATCC). LNCaP cells were cultured in growth medium (phenol-red-free RPMI 1640 medium (Invitrogen, 11835-063) containing 2 mM L-glutamine (Invitrogen, 25030-024) and 1% (vol/vol) penicillin–streptomycin (10,000 units/ml penicillin and 10,000 μg/ml of streptomycin utilizing penicillin G sodium salt) and streptomycin sulfate (prepared in normal saline; Invitrogen, 15140122) and 10% (vol/vol) fetal bovine serum (FBS)) in a 5% CO2 air incubator at 37 °C. We collected cells for assay from T175 stock flasks by washing them once in PBS (pH 7.4) (Invitrogen, 14190-094) and harvested them using 5 mL of 1× trypsin–EDTA (10× trypsin–EDTA, 5.0 g/L trypsin, 2.0 g/L of EDTA4Na and 8.5 g/L of NaCl, without phenol red; Invitrogen, 15400-054) diluted in PBS solution. A 5-mLvolume of assay medium (the same as growth medium except that 5% (vol/vol) charcoal-stripped FBS (HyClone, SH30068.03) was included instead of 10% (vol/vol) FBS) was then added to each flask. Cells were syringed at least twice using a sterile 18-gauge 1.5-inch (1.2 × 40 mm) broad-gauge needle, and cell density was measured using a Countess automated cell counter (Life Technologies). Cells were further diluted in growth medium plus 5% (vol/vol) charcoal-stripped FBS and seeded at a density of 6.5 × 103 cells per well (in 90 μL) into transparent, black, tissue-culture-treated 96-well plates (Corning, 3904). Plates were incubated overnight at 37 °C, 5% CO2. A 10× compound dose-response plate with DMSO normalized to 3% (vol/vol) was generated in 30 μL of assay media using the Labcyte Echo 555 acoustic dispenser, and a 10-μL volume of diluted compound was then dosed to cells using a Thermo Scientific Matrix PlateMate to give a final concentration of 0.3% (vol/vol) DMSO and 8- or 12-point half-log singlicate concentration range (30 μM or 3 μM top concentration). DMSO was dispensed in the maximum-signal control wells to give a final concentration of 0.3% (vol/vol), and either AZ13182659 or 6 was dispensed to generate a minimum signal to give a final concentration of 30 μM or 3 μM, respectively. Plates were then further incubated for 20–22 h at 37 °C, 5% CO2. Plates were then fixed by the addition of 20 μL of 10% (vol/vol) formaldehyde solution (in PBS) to each well (final formaldehyde concentration: 1.67% (vol/vol)) and left at room temperature for 10 min. The fixative solution was removed and cells were washed three times with 250 μL of PBST using a Tecan PW96 plate washer (Tecan UK), and 50 μl of PBS was added to all wells. Immunostaining was performed at room temperature. Cells were permeabilized by the addition of 35 μL of PBS containing 0.5% (vol/vol) Tween 20 and incubated for 1 h at room temperature. Permeabilization solution was removed and cells were washed three times with 250 μL of PBST using an automated plate washer. 35 μL of blocking solution (PBST containing 3% (wt/vol) Marvel dried skimmed milk (Nestle)) was added to each well and plates were incubated at room temperature for a minimum of 1 h. After removal of the blocking solution with a plate washer, 35 μL of monoclonal mouse anti-human AR (clone AR441; DAKO, M3562), diluted 1:500 in blocking solution, was added to each well and incubated for 1 h.
The primary antibody solution was removed from the wells, and wells were then washed three times with 100 μL of PBST each time using a plate washer. Then 35 μL of secondary Alexa Fluor 488–conjugated goat anti-mouse IgG (Invitrogen, A-11001), diluted 1:500, and Hoechst (Molecular Probes), diluted 1:5,000 in blocking solution, was added to each well. From that point on, whenever possible, plates were protected from light exposure. The plates were incubated for 1 h. Then the secondary antibody solution was removed from the wells and the wells were washed three times with 100 μL of PBST each time using a plate washer. Then 50 μL of PBST was added to each well and plates were covered with a black plate seal. Cell images were acquired using a Cellomics Cell Insight reader (Thermo Scientific) using a 10×/0.3-NA objective and applying the filters BGRFR_386_23 and BGRFR_485_20 to detect Hoechst and Alexa Fluor 488 signals. Using the Compartmental Analysis v4 bio-application, we generated nuclear masks from the Hoechst-stained nuclei in channel 1 and applied a suitable threshold in channel 2 to quantify AR within the nuclear masks. The mean total intensity was normalized for cell number, giving the total intensity of AR staining per cell. Sample images are provided in Supplementary Figure 15.
384-well method using rabbit anti-human AR.
LNCaP cells were cultured as described above. Cells were seeded into black, clear-bottom, 384-well plates (Costar, 3712) at 3,000 cells per well in a volume of 40 μl using a Multidrop Combi dispenser (Thermo Scientific) and then incubated overnight at 37 °C, 5% CO2. An Echo 555 acoustic dispenser was used to dispense test compounds directly into cell plates, producing a 12-point half-log singlicate concentration range (30 μM or 3 μM top concentration). Minimum and maximum controls were the same as for the 96-well assay. Plates were then further incubated with compound for 18 h at 37 °C, 5% CO2. Cells were fixed by the addition of 20 μl/well of 12% (vol/vol) formaldehyde using a Thermo Scientific Matrix WellMate liquid handler (final formaldehyde concentration: 4% (vol/vol)) and plates were incubated at room temperature for 20 min. The fixative solution was removed and cells were washed once with 250 μL of PBST using a BioTek ELx405 plate washer (BioTek Instruments, UK). Cells were then permeabilized by 20 μl/well PBS containing 0.1% Triton X-100 and incubated for 20 min at room temperature. Cells were washed once in PBS. Next, 20 μl/well of primary rabbit anti-human AR (PG-21; Millipore, 06-680) diluted 1:400 in antibody buffer (PBST + 5% FCS) was added per well and plates were incubated overnight at 4 °C. The following day, the plates were washed three times with PBS before the addition of 20 μl/well secondary antibody (goat anti-rabbit–Alexa Fluor 488 (A31627; Molecular Probes, Life Technologies, Paisley, UK) diluted 1:500 in antibody buffer plus Hoechst nuclear stain diluted 1:5,000). Plates were protected from light and incubated for 1 h at room temperature. The plates were then washed three times in PBS without a final aspiration step and covered with black plate seals. Cell images were acquired as above for the 96-well method. Sample images are provided in Supplementary Figure 16.
Data analysis.
During the routine running of the biochemical and cellular cascade, experimental data from each of the assays were exported into Genedata Screener client graphing software (Genedata AG, Basel, Switzerland) for curve-fitting analysis. For the representative curves exemplified in the figures, the data were plotted using Prism software (GraphPad Prism 6, La Jolla, CA). A nonlinear regression curve fit using four parameters with variable slope was applied.
We determined IC50 values by calculating the concentration of test compound required to give a 50% reduction of the total measured effect corrected for background (the average maximum control value minus the average minimum control value). The quality of each assay was determined by analysis of the Z factor50.
Measurement of AR binding.
Binding to AR was measured in LNCaP cellular extracts according to an established method51.
BROMOscan profiling.
BROMOscan is a technology platform for identifying small-molecule BRD inhibitors. Based on proven KINOMEscan technology, BROMOscan uses a proprietary ligand-binding-site-directed competition assay to quantitatively measure interactions between test compounds and BRDs. This robust and reliable assay panel is suitable for high-throughput screening and delivers quantitative ligand-binding data to facilitate the identification and optimization of potent and selective small-molecule BRD inhibitors. BROMOscan assays include trace BRD concentrations (<0.1 nM) and thereby report true thermodynamic inhibitor Kd values over a broad range of affinities (<0.1 nM to >10 μM).
BROMOscan primary screen protocol.
T7 phage strains with BRDs were grown in parallel in 24-well blocks in an E. coli host derived from the BL21 strain. E. coli were grown to log phase and infected with T7 phage from a frozen stock (multiplicity of infection: 0.4) and incubated with shaking at 32 °C until lysis (90–150 min). The lysates were centrifuged (5,000g) and filtered (0.2 μm) to remove cell debris. Streptavidin-coated magnetic beads were treated with biotinylated small-molecule or acetylated peptide ligands for 30 min at room temperature to generate affinity resins for BRD assays. The liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1% BSA, 0.05% Tween 20, 1 mM DTT) to remove unbound ligand and to reduce nonspecific phage binding. We assembled binding reactions by combining BRDs, liganded affinity beads and test compounds in 1× binding buffer (16% SeaBlock, 0.5× PBS, 0.02% BSA, 0.04% Tween 20, 0.004% sodium azide, 7.9 mM DTT). Test compounds were prepared as 1,000× stocks in 100% DMSO and subsequently diluted 1:25 in monoethylene glycol (MEG). The compounds were then diluted directly into the assays such that the final concentrations of DMSO and MEG were 0.1% and 2.4%, respectively. In the primary screen compounds were tested at a final concentration of 3 μM. All reactions were carried out in polypropylene 384-well plates in a final volume of 0.02 ml. The assay plates were incubated at room temperature with shaking for 1 h, and the affinity beads were washed with PBST. The beads were then resuspended in elution buffer (PBST, 2 μM nonbiotinylated affinity ligand) and incubated at room temperature with shaking for 30 min. The BRD concentration in the eluates was measured by qPCR.
BROMOscan Kd protocol.
This was carried out as in the primary screen protocol described above, except for compound handling. An 11-point threefold serial dilution of each test compound was prepared in 100% DMSO at a 1,000× final test concentration. This serial was then diluted to 100× in ethylene glycol and subsequently diluted to 1× in the assay (final DMSO concentration, 0.1%; ethylene glycol concentration, 0.9%). Kd values were determined using a compound top concentration of 10 μM. Binding constants (Kd) were calculated with a standard dose-response curve using the Hill equation:

The Hill slope was set to −1. Curves were fitted using a nonlinear least-squares fit with the Levenberg–Marquardt algorithm.
Computational modeling.
Predicted structure models of the in cis and in trans complexes were built from the crystal structure of 3. BD2 was constructed through homology with the second BD1 of the crystal structure, and the interdomain region was constructed using the MOE homology model builder (Chemical Computing Group, Montreal, QC, Canada) with default parameters. The relative positioning of the two pairs of domains for the in trans model was chosen so as to place the relevant termini approximately as far apart as they appear in the in cis model.
NMR spectroscopy.
All NMR experiments were performed at 30 °C on a Bruker Avance III 800 MHz (18.8 T) spectrometer running TopSpin v3.2 and equipped with a 5-mm z-gradient 1H/13C/15N TCI probe. The NMR sample used for the titrations contained 0.03 mM of 6 or 0.14 mM of 1, uniformly 2H,15N-labeled BRD4(44–460) in 20 mM Na2HPO4, 2 mM TCEP (tris(2-carboxyethyl)phosphine), and 7%/93% D2O/H2O at pH 6.7. Unlabeled 1 and 6 were dissolved in DMSO to give a final concentration of 100 mM and were subsequently titrated into the protein sample to give ligand:protein stoichiometries of 0:1, 1:1 and 2:1. Data sets were processed using the program nmrPipe52 and plotted using the Python program nmrGlue53.
We distinguished between the two BRDs through comparison with 2D TROSY HSQC spectra of isolated BRD4(1) and BRD4(2). BD1 has three tryptophans in its sequence, whereas BD2 contains one (W374), with indole nitrogen 1H resonances located between 9.6 and 10.5 p.p.m. Hence, by comparing the spectra of isolated BRD4(2) and BRD4(1,2) (Supplementary Fig. 7b), we were able to directly assign the signal for W374.
Analytical ultracentrifugation.
Protein samples of BRD4(40–169) and BRD4(1–477) were characterized by sedimentation velocity analytical ultracentrifugation. Samples of protein of ∼50 μM concentration were initially analyzed and buffer-exchanged on a Superdex200 gel filtration column (GE) coupled to a Wyatt multi-angle light-scattering detector, a refractive index detector and a UV detector, which was equilibrated in 10 mM Tris, pH 7.4, 150 mM NaCl. This performed buffer exchange and allowed analysis of sample heterogeneity and mass.
All compounds were separately dissolved to a concentration of 1 mM in 100% DMSO. Protein samples were diluted in 10 mM HEPES, pH 7.4, 150 mM NaCl to final concentrations of ∼20 μM for BRD4(40–169) and 8 or 11 μM for BRD4(1–477). The final concentration of compounds was adjusted to compound:protein molar ratios of 2:1, 1:1, 0.5:1 and 0:1 by addition to the above-mentioned protein samples.
400 μL of each of the above-mentioned samples was loaded into two-sector epon centerpieces with sapphire glass and centrifuged at 50,000 r.p.m. in a Beckman XL-I ultracentrifuge at 20 °C, with scans collected every 1 min until sedimentation of the protein was reached. Data were analyzed using the sedimentation distribution modeling software Sedfit (REF) and were used to calculate the frictional ratio (f/f0) from the frictional coefficient (f) and that of a sphere (f0). Analysis of continuous c(s, f/f0) contour analysis was performed in Sedfit.
Small-angle X-ray scattering.
The synchrotron radiation X-ray scattering data from solutions of truncated BRD4 construct containing two BRDs were collected in two experimental sessions at beamline BM29 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France)54. With the use of a Pilatus 1M detector at a sample-to-detector distance of 2.867 m and a wavelength of λ = 0.09919 nm, the range of momentum transfer 0.05 < s < 5.0 nm−1 was covered (s = 4πsinθ/λ, where 2θ is the scattering angle). The apo protein solutions and those containing test compounds were measured at concentrations of 1, 2, 5 and 10 mg/ml using a continuous flow cell capillary. To monitor for the radiation damage, we compared 50 successive 0.2-s exposures of protein solutions; frames with statistically significant changes were discarded. The data were normalized to the intensity of the transmitted beam and radially averaged; the scattering of the buffer was subtracted and the difference curves were scaled for protein concentration. The low-angle data measured at lower protein concentrations were extrapolated to infinite dilution and merged with the highest concentration data to yield the final composite scattering curves.
The forward scattering I(0), the radius of gyration Rg, the distance distribution function of the particle p(r) and the maximum dimension Dmax were derived using the automated SAXS data analysis pipeline55. The p(r) functions were further recomputed manually with the program GNOM56. The molecular masses were evaluated by comparison of the forward scattering with that from a BSA (66-kDa molecular weight) reference solution.
Ab initio shape models of the BRD4 constructs were generated using the bead modeling program DAMMIN57. This program represents the particle shape by an assembly of densely packed beads and uses simulated annealing to construct a compact interconnected model fitting the experimental data. Multiple runs were performed to generate 15–20 models.
Using the Ensemble Optimization Method (EOM)22, we generated a pool of 10,000 models comprising two rigid BRDs (residues 42–168, PDB ID 2OSS, and 349–458, PDB ID 2YEM) connected by a flexible linker, N-terminal tag and flexible N and C termini. The scattering from each model from the pool was calculated with the program CRYSOL58. A subset of the pool was selected using a genetic algorithm such that the calculated averaged scattering of the selected models agreed with the experimental data. We obtained the Rg distributions of the selected ensembles by repeating the selection process multiple times. For the 100 models best fitting the experimental scattering from each construct, the histograms of the distances between the centers of the two BRDs were computed. For the top-fitting model for each construct, we computed the sedimentation coefficients using WinHydroPro v.1.00 (ref. 59).
1% DMSO was added to the inhibitor-containing solutions to improve their solubility. To verify that DMSO did not influence the results, we measured the apo protein in 0%, 1% and 10% DMSO; after subtraction of the scattering of the appropriate DMSO-containing buffers, we found that DMSO addition did not affect the SAXS profile.
All samples were measured at solute concentrations of 1, 2, 5 and 10 mg/ml. Concentration dependence was observed for all samples, indicating increased protein aggregation or oligomerization with concentration. The magnitude of the concentration effect was different for the different constructs. For the apo protein data (Supplementary Fig. 17a) the effect was moderate, indicating some degree of oligomerization (quantitatively, moderate dimerization). A similar albeit somewhat larger effect was observed in the presence of 1. For 6 (Supplementary Fig. 17b), the concentration effect was much more pronounced, indicating formation of substantial amounts of larger oligomers (tetra-, octa- and even larger). The data from all these concentration series were extrapolated to infinite dilution, and further analysis for all constructs was performed on the extrapolated data, which are expected to correspond to monomeric species (Supplementary Fig. 18). The p(r) function of the apo protein has a skewed shape typical for very elongated structures, with the maximum corresponding to the particle cross-section (about 4 nm). From experimental data from the apo protein and from the mixtures with the ligands 1 and 6, each at three stoichiometries (ligand:protein ratios of 0.5:1, 1:1 and 2:1), extrapolated to infinite dilution, distance distribution functions were calculated using GNOM56. The overall structural parameters apparent molecular mass computed from forward scattering, radius of gyration (Rg) and maximum particle dimension (Dmax) were extracted from the extrapolated curves (Supplementary Table 8).
A further and more detailed analysis of the results was carried out using BRD4 sequence information. The available crystal structures of the BRDs (PDB IDs 2OSS and 2YEM) correspond to residues 42–168 and 349–458 in the full-length construct, which consists of His-tag (24 amino acids (aa)) + N terminus (41 aa), BD1 (127 aa), a 180-aa-long interdomain linker, BD2 (111 aa) and the C terminus (18 aa).
According to the NORSnet prediction60, residues 1–60 and 168–351 are likely to be disordered. Therefore, it seemed likely that BRD4 was flexible in solution and that appropriate analysis methods should be used to account for that. We used the EOM, a technique whereby a large pool of randomized models is generated and sub-ensembles are selected in such a way that their mixtures fit the experimental data22. We generated 10,000 models, keeping the crystal structures of the domains intact and randomizing the linkers. The properties of the system could be inferred from the analysis of the distributions of the overall parameters (for example, Rg and interdomain distance) in the selected ensembles compared with the random pool.
BRET and NanoBRET assay.
Studies were carried out with HEK293 or HCT116 cells. HEK293 human embryonic kidney cells (ATCC; CCL-247) were cultured in DMEM containing 2 mM glutamine and 10% FCS. HCT116 human colorectal carcinoma cells (ATCC; CCL-1573) were cultured in McCoy's 5A medium containing 2 mM glutamine and 10% FCS. Cells (8 × 105) were seeded into wells of a six-well culture plate (Corning) and allowed to attach for 6 h. They were then co-transfected with Histone H3.3-HaloTag (NM_002107) and NanoLuc-BRD4 full-length (1–1,362) (060885), NanoLuc-BRD4 full-length N433F mutant, or NanoLuc-N-terminal BRD4-BD1 (aa 44–168) expression vectors (Promega). For intramolecular biosensor experiments, cells were transfected with a HaloTag-BRD4-BD1_BD2-NanoLuc (aa 44–460) fusion or HaloTag-BRD4-BD1(N140F)_BD2-NanoLuc mutant fusion expression vectors (Promega).
Twenty hours after transfection, cells were harvested and resupended in OptiMEM (Life Technologies) supplemented with 4% FCS at a density of 2 × 105 cells/ml in the absence (control) or presence (experimental) of 100 nM HaloTag NanoBRET 618 fluorescent ligand (Promega). Cells were then seeded into white, flat-bottomed tissue-culture-treated plates (Greiner), with 90 μl of cell suspension per well, to which test compounds or vehicle (final DMSO concentration: 0.1% (vol/vol)) were added as tenfold solutions in complete media at various concentrations. Plates were incubated for 18 h at 37 °C in the presence of 5% CO2. NanoBRET Nano-Glo Substrate (Promega) was added to both control and experimental samples at a final concentration of 10 μM. Plates were read within 10 min using a CLARIOstar plate reader (BMG Labtech) equipped with a 450/80-nm bandpass and 610-nm longpass filter module. A corrected BRET ratio was calculated, defined as the ratio of the emission at 610 nm/450 nm for experimental samples (i.e., those treated with HaloTag NanoBRET 618 fluorescent ligand) minus the emission at 610 nm/450 nm for control samples (i.e., those not treated with HaloTag NanoBRET 618 fluorescent ligand). BRET ratios were expressed as milliBRET units (mBU), where 1 mBU corresponds to the corrected BRET ratio multiplied by 1,000.
Cell transfection, treatments and quantitative NanoBRET target engagement measurements under equilibrium conditions.
Target engagement at NanoLuc-BRD4 was performed as described previously26. To determine unlabeled compound isotherms, we added the iBET-NCT tracer to the cells at a fixed concentration of 0.2 μM before test compound addition. Serially diluted test compounds were then added to the cells and allowed to equilibrate for 3 h. For BRET measurements, NanoBRET NanoGlo Substrate (Promega) was added to the cells, and filtered luminescence was measure on a Promega Glomax Discover luminometer equipped with 450-nm bandpass filter (donor) and 610-nm longpass filter (acceptor) using a 0.5-s integration time. Competitive displacement data were then graphed with GraphPad Prism software using a three-parameter curve fit with the following equation:

We generated normalized data by assigning 100% to the maximum of the three-parameter curve fit and 0% for the theoretical minimum value of the three-parameter curve fit.

Kinetic analysis of intracellular residence time at BRD4 via NanoBRET.
For direct analysis of relative compound dissociation rates via BRET, 2 × 106 cells transfected with NLuc-BRD4 constructs were processed as described previously26. Briefly, transfected cells were first pre-equilibrated with test compound for 3 h at 37 °C, 5% CO2. Cells treated with compound were pelleted, washed once with PBS, and resuspended in 10 mL of Opti-MEM in the presence of NanoBRET NanoGlo Substrate and 1 μM iBET-NCT tracer immediately before BRET measurements. BRET data were normalized to 100% signal from the positive control (defined as the maximum BRET value observed from cells treated with 1 μM iBET-NCT in the absence of unlabeled competing compound) and 0% signal (defined as the BRET value observed from cells treated with 1 μM iBET-NCT + 10 μM unmodified 1).
Cell lines.
All cell lines were originally purchased from ATCC or DSMZ, cultured according to the vendors' instructions, authenticated using DNA fingerprinting short-tandem-repeat assay and determined by testing to be free of mycoplasma contamination. All revived cells were used within 15 passages and less than 6 months.
Immunofluorescent foci assays for BRD4, MED1 and H3K27Ac.
U2OS cells (ATCC) were plated in 96-well plates at 16,000 cells per well. Cells were incubated at 37 °C in 5% CO2 overnight and then dosed with compounds for 4 h. DMSO was used as a maximum control. Cells were fixed by 4% formaldehyde at room temperature (RT) for 10 min and subjected to 0.1% saponin permeabilization for another 10 min. Cells were co-stained with anti-BRD4 (1:25, Sigma-Aldrich, HPA015055-100UL) with either anti-MED1 (1:50, Bethyl Laboratories, A303-876A) or anti-H3K27Ac (1:250, Diagenode, C15200184) for 2 h at 37 °C, washed with PBS four times, and labeled by Alexa Fluor 488–conjugated goat anti-rabbit (Life Technologies; A11034) with either Alexa Fluor 594–conjugated donkey anti-goat (Life Technologies; A11058) or Alexa Fluor 594–conjugated goat anti-mouse at 1:200 dilution (Life Technologies; A11032) for 1 h at RT. After being washed with PBS twice, cells were stained with Hoeschst at 1:4,000 (Molecular Probes, Life Technologies, Paisley, UK; H21492) for 10 min, washed twice with PBS and then subjected to image acquisition by the ImageXpress MicroXL High Content Screening System (Molecular Devices). The data were analyzed by MetaXpress (Molecular Devices). IC50 was calculated using GraphPad Prism.
c-Myc flow assay.
MM.1S cells (ATCC) were plated in 96-well plates at 200,000 cells per well, treated with serial concentrations of compound 1 h after plating and grown for 16 h at 37 °C. Cells were transferred into a 96-well V-bottom plate and fixed sequentially with 2% paraformaldehyde for 10 min at 37 °C followed with 90% cold methanol for 10 min on ice. Cells were washed and blocked in incubation buffer (PBS + 0.5% BSA) for 10 min and incubated with anti–c-Myc as a primary antibody (1:200, Cell Signaling Technology, 5605) at RT for 1 h. Cells were washed twice and then stained with Alexa Fluor 488–conjugated anti-rabbit IgG (1:1,000, Cell Signaling Technology, 4412) for 30 min at RT and washed again. The pellet was resuspended in incubation buffer and analyzed by flow cytometer (BD FACSCalibur) using the FL-1 detector. Data were analyzed using FlowJo software V7. We selected the live cell population by gating on forward scatter/side scatter, and we used the Geo Mean fluorescence of FL-1 signal to calculate IC50 values.
Western blotting experiments.
4 × 106 MM.1S cells were seeded in each well of the six-well plates and treated with compounds at various concentrations for 4 h. Crude lysate was prepared with 1× SDS lysis buffer (0.06 M Tris-HCl, pH 6.8, 1% SDS and 10% glycerol). Samples with 20 μg of total protein were loaded and western blots were run using typical procedures. We used primary c-Myc antibody (CST, 5605) at 1:1,000 dilution, GAPDH antibody (CST, 5174) at 1:10,000 dilution, and secondary anti-rabbit HRP-conjugated antibody (CST; 7074) at 1:10,000 dilution. The protein level was visualized using the ImageQuant LAS4000.
Cell growth inhibition and viability assays.
Optimal 384-well seeding densities were pre-measured for linear growth over 72 h. 30 μl of cells per well were seeded in black, flat-bottom, 384-well plates and treated with compounds for 72 h. The viable cells were measured using Alamar Blue reagents according to the manufacture's protocol (Invitrogen), and the percentage of viable cells was calculated by comparison to no-drug controls. We calculated the percent net growth by normalizing against the no-drug controls, and the GI50 value was calculated using GraphPad Prism 6.
The viability assay was performed according to the same protocol used for the growth inhibition assay but differed in data treatment. The absolute number of remaining viable cells, instead of the percent net growth, was determined after 72 h of compound treatment. Dose-response curves were plotted in GraphPad Prism and fitted using sigmoidal nonlinear regression to determine IC50 values.
Gene expression analysis by RNA-seq.
To compare the genome-wide transcriptional consequences induced by the different BET inhibitors, we carried out RNA-seq on hematologic cancer cell lines (K-562, MM.1S and MV-4-11, obtained from ATCC) after treatment with 1 (2 μM), 6 (0.05 μM) or DMSO control for 24 h. Correlation analysis of the gene expression changes after inhibitor treatment showed that the gene abundance differential was highly correlated among all compounds, indicating that the transcription profiles of all inhibitors were similar.
After BET inhibitor (6, 0.05 μM; 1, 2 μM,) or DMSO vehicle treatment of cell lines (MV-4-11, MM.1S and K-562) for 24 h, we carried out RNA isolation, cDNA library generation and sequencing to 12 million reads on the Illumina HighSeq at Q2 Solutions (http://www.q2labsolutions.com/). Bcbio-nextgen (https://github.com/chapmanb/bcbio-nextgen) was used to process FastQ files, perform quality control, align to genome build hg38 using HISAT2 (ref. 61), and quantify transcription expression based on Ensembl annotation. DESeq2 (ref. 62) version 1.12.1 was used to identify differentially expressed genes and calculate log10 fold change values (Supplementary Fig. 19).
Accession codes.
PDB: 5AD2 for BRD4(1) in complex with 2; 5AD3 for BRD4(1) in complex with 6.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper. Reprints and permissions information is available online at http://www.nature.com/reprints/index.html. Correspondence and requests for materials should be addressed to M.J.W. or E.C.
References
Vidler, L.R., Brown, N., Knapp, S. & Hoelder, S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 55, 7346–7359 (2012).
Prinjha, R.K., Witherington, J. & Lee, K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 33, 146–153 (2012).
Nicodeme, E. et al. Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123 (2010).
Mirguet, O. et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 56, 7501–7515 (2013).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Wu, S.-Y. & Chiang, C.-M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 282, 13141–13145 (2007).
Vollmuth, F., Blankenfeldt, W. & Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 284, 36547–36556 (2009).
Delmore, J.E. et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011).
Dawson, M.A. et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478, 529–533 (2011).
Stathis, A. et al. Results of a first-in-man phase I trial assessing OTX015, an orally available BET-bromodomain (BRD) inhibitor, in advanced hematologic malignancies. Eur. J. Cancer 50, 196 (2014).
Bradbury, R.H. et al. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg. Med. Chem. Lett. 23, 1945–1948 (2013).
Loddick, S.A. et al. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol. Cancer Ther. 12, 1715–1727 (2013).
Quinn, E. et al. BROMOscan—a high throughput, quantitative ligand binding platform identifies best-in-class bromodomain inhibitors from a screen of mature compounds targeting other protein classes. Cancer Res. 73, 4238 (2013).
Morris, J. et al. NCI-60 response profiles of >400 investigational oncology agents: a resource enabling drug and biomarker discovery. Mol. Cancer Ther. 12, A102 (2013).
Morris, J. et al. Interrogation of NCI-60 patterns of growth inhibition in conjunction with investigational oncology agents kinase profiling for the elucidation of mechanistic targets. Cancer Res. 74, 5475 (2014).
Asangani, I.A. et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 510, 278–282 (2014).
Feng, Q. et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 24, 809–819 (2014).
Williams, F.P., Milbradt, A.G., Embrey, K.J. & Bobby, R. Segmental isotope labelling of an individual bromodomain of a tandem domain BRD4 using sortase A. PLoS One 11, e0154607 (2016).
Lebowitz, J., Lewis, M.S. & Schuck, P. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 (2002).
Brown, P.H. & Schuck, P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 90, 4651–4661 (2006).
Mertens, H.D. & Svergun, D.I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 172, 128–141 (2010).
Tria, G., Mertens, H.D., Kachala, M. & Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 (2015).
Machleidt, T. et al. NanoBRET—a novel BRET platform for the analysis of protein–protein interactions. ACS Chem. Biol. 10, 1797–1804 (2015).
Baud, M.G.J. et al. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 346, 638–641 (2014).
Philpott, M. et al. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenetics Chromatin 7, 14–25 (2014).
Robers, M.B. et al. Target engagement and drug residence time can be observed in living cells with BRET. Nat. Commun. 6, 10091 (2015).
Whyte, W.A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Creyghton, M.P. et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936 (2010).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Arrowsmith, C.H. et al. The promise and peril of chemical probes. Nat. Chem. Biol. 11, 536–541 (2015).
Bradbury, R.H. et al. Optimization of a series of bivalent triazolopyridazine based bromodomain and extraterminal inhibitors: the discovery of (3R)-4-[2-[4-[1-(3-methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153). J. Med. Chem. 59, 7801–7817 (2016).
Chen, H. et al. AZD5153: a novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol. Cancer Ther. http://dx.doi.org/10.1158/1535-7163.MCT-16-0141 (2016).
Krishnamurthy, V.M., Estroff, L.A. & Whitesides, G.M. Multivalency in ligand design. in Fragment-based Approaches in Drug Discovery (eds. Jahnke, W. & Erlanson, D.A.) Ch. 2 (Wiley, Weinheim, Germany, 2006).
Arnold, D.L., Foreman, K.W., Jin, M., Wanner, J. & Werner, D.S. Preparation of bivalent bromodomain ligands for treating a disease associated with a protein having tandem bromodomains. International patent application WO2013033268 A2 (2013).
Arnold, D.L., Foreman, K.W. & Werner, D.S. Preparation of bivalent bromodomain ligands, and methods of using the same. International patent application WO2015081284 (2015).
Sun, H. et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J. Am. Chem. Soc. 129, 15279–15294 (2007).
Jacobson, R.H., Ladurner, A.G., King, D.S. & Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 288, 1422–1425 (2000).
Ruthenburg, A.J., Li, H., Patel, D.J. & Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994 (2007).
Morinière, J. et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 461, 664–668 (2009).
Ruthenburg, A.J. et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145, 692–706 (2011).
Buttar, D. et al. A combined spectroscopic and crystallographic approach to probing drug-human serum albumin interactions. Bioorg. Med. Chem. 18, 7486–7496 (2010).
Camenisch, G. et al. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs' lipophilicity and molecular weight. Eur. J. Pharm. Sci. 6, 313–319 (1998).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A.J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W.G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Bricogne, G. et al. Buster Version 2.11.6. (Global Phasing Ltd., 2011).
Smart, O.S. et al. Grade Version 1.2.9. (Global Phasing Ltd., 2011).
Callis, R. et al. A screening assay cascade to identify and characterize novel selective estrogen receptor downregulators (SERDs). J. Biomol. Screen. 20, 748–759 (2015).
Zhang, J.H., Chung, T.D. & Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 (1999).
Zava, D.T., Landrum, B., Horwitz, K.B. & McGuire, W.L. Androgen receptor assay with [3H]methyltrienolone (R1881) in the presence of progesterone receptors. Endocrinology 104, 1007–1012 (1979).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Helmus, J.J. & Jaroniec, C.P. Nmrglue: an open source Python package for the analysis of multidimensional NMR data. J. Biomol. NMR 55, 355–367 (2013).
Pernot, P. et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 (2013).
Franke, D., Kikhney, A.G. & Svergun, D.I. Automated acquisition and analysis of small angle X-ray scattering data. Nuc. Inst. Meth. A. 689, 52–59 (2012).
Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 (1992).
Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 (1999).
Svergun, D.I., Barberato, C. & Koch, M.H.J. CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 (1995).
Ortega, A., Amorós, D. & García de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 (2011).
Schlessinger, A., Liu, J. & Rost, B. Natively unstructured loops differ from other loops. PLoS Comput. Biol. 3, e140 (2007).
Kim, D., Langmead, B. & Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Love, M.I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
The authors thank G. Carr, S. Lamont, T. Moss, A. Patel and G. Wrigley for the synthesis of test compounds; H. Ashton for coordinating biological testing at DiscoverX; K. Eckersley and P. McArdle for supporting cellular imaging studies; and P. Filippakopoulos for BRD4 mutant vector constructs. We thank K. Embrey for helpful comments on the compilation of the manuscript.
Author information
Authors and Affiliations
Contributions
M.J.W. designed compounds, coordinated chemistry, analyzed and interpreted data and coordinated manuscript preparation. H.C. coordinated cell biology work and analyzed and interpreted biological and biophysical data. A.A.R. designed compounds, carried out computational modeling and structural analysis and interpreted data. G.W. coordinated biophysical testing and AR/ER-based cell experiments and conceived of and coordinated NanoBRET experiments. R.B. designed and carried out NMR studies. S.B. carried out cell biology experiments. R.H.B. designed compounds, coordinated chemistry and analyzed and interpreted data. R.C. carried out ER cell biology experiments. E.C. was the project leader. I.D. carried out NanoBRET experiments. D.L.D. coordinated NanoBRET target engagement experiments and construct production. A.D. coordinated transciptomic profiling and interpreted data. L.F. designed, expressed and purified protein constructs. G.H. analyzed and interpreted biophysical data. T.A.J. carried out AUC experiments and ab initio modeling. A.K. carried out SAXS experiments. M.M. coordinated AUC experiments and analyzed and interpreted biophysical data. J.M. conducted NanoBRET target engagement experiments. D.O. coordinated SAXS experiments and analyzed and interpreted biophysical data. J.P. carried out protein X-ray structure determination. P.P. carried out the c-Myc flow cell assay. G.R.R. carried out computational modeling of tandem domain proteins. M.B.R. carried out NanoBRET target engagement experiments. S.S. carried out transcriptomics experments. N.S. carried out AR cell biology experiments. D.I.S. coordinated and carried out SAXS experiments. W.W. carried out cell biology experiments. D.W. synthesized compounds. D.M.W. analyzed and interpreted the data. Y.Y. carried out transcriptomics experiments. All authors discussed results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The work described here was initiated as a project within AstraZeneca. M.J.W., A.A.R., G.W., R.H.B., D.O. and J.P. are former employees of AstraZeneca and may hold stock in the company. H.C., R.B., S.B., R.C., E.C., I.D., A.D., L.F., G.H., M.M., P.P., G.R.R., S.S., N.S., W.W., D.W., D.M.W. and Y.Y. are current employees of AstraZeneca and may hold stock in the company. Promega Corp. carried out the NanoBRET experiments as part of a collaboration with AstraZeneca. D.L.D., J.M. and M.B.R. are employees of Promega Corp. and may hold stock in the company. The University of Manchester carried out the AUC studies at the request of AstraZeneca. T.A.J. is an employee of the University of Manchester. The European Molecular Biology Laboratory carried out the SAXS studies at the request of AstraZeneca. A.K. and D.I.S. are employees of the European Molecular Biology Laboratory.
Supplementary information
Supplementary Text and Figures
Supplementary Results, Supplementary Tables 1–10, Supplementary Figures 1–19 and Supplementary Note 2. (PDF 3096 kb)
Supplementary Note 1
Synthetic Procedures. (PDF 364 kb)
Rights and permissions
About this article

Cite this article
Waring, M., Chen, H., Rabow, A. et al. Potent and selective bivalent inhibitors of BET bromodomains. Nat Chem Biol 12, 1097–1104 (2016). https://doi.org/10.1038/nchembio.2210
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nchembio.2210
This article is cited by
-
Targeting bromodomain-containing proteins: research advances of drug discovery
Molecular Biomedicine (2023)
-
Effects of bromodomain and extra-terminal inhibitor JQ1 and interleukin-6 on breast cancer cells
Molecular Biology Reports (2023)
-
Phosphorylation-dependent BRD4 dimerization and implications for therapeutic inhibition of BET family proteins
Communications Biology (2021)
-
Trivalent PROTACs enhance protein degradation via combined avidity and cooperativity
Nature Chemical Biology (2021)
-
Enhancer rewiring in tumors: an opportunity for therapeutic intervention
Oncogene (2021)
