
Abstract
Depression is the most prevalent psychiatric disorder with a complex and elusive etiology that is moderately heritable. Identification of genes would greatly facilitate the elucidation of the biological mechanisms underlying depression, however, its complex etiology has proved to be a major bottleneck in the identification of its genetic risk factors, especially in genome-wide association-like studies. In this study, we exploit the properties of a genetic isolate and its family-based structure to explore whether relatively rare exonic variants influence the burden of depressive symptoms in families. Using a multistep approach involving linkage and haplotype analyses followed by exome sequencing in the Erasmus Rucphen Family (ERF) study, we identified a rare (minor allele frequency (MAF)=1%) missense c.1114C>T mutation (rs115482041) in the RCL1 gene segregating with depression across multiple generations. Rs115482041 showed significant association with depressive symptoms (N=2393, βT-allele=2.33, P-value=1 × 10−4) and explained 2.9% of the estimated genetic variance of depressive symptoms (22%) in ERF. Despite being twice as rare (MAF<0.5%), c.1114C>T showed similar effect and significant association with depressive symptoms in samples from the independent population-based Rotterdam study (N=1604, βT-allele=3.60, P-value=3 × 10−2). A comparison of RCL1 expression in human and mouse brain revealed a striking co-localization of RCL1 with the layer 1 interlaminar subclass of astrocytes found exclusively in higher-order primates. Our findings identify RCL1 as a novel candidate gene for depression and offer insights into mechanisms through which RCL1 may be relevant for depression.
Similar content being viewed by others

Introduction
Depression is a psychiatric illness that is moderately heritable (h2=40–50%)1 and has a high prevalence (8.6–17.1%).2, 3, 4 Although all earlier genome-wide association studies (GWAS) failed to identify genes underlying depression in European populations,5, 6, 7, 8, 9, 10 17 common genetic variants were recently identified only after increasing the sample size to over 75 000 cases and 230 000 controls11 in the GWAS. All identified variants are non-coding with modest effects on depression (odds ratio<1.05) and together explain a small percentage of variation in depression.11 Although expanding array-based GWAS further may deliver more common genetic variants associated with depression, the effects of these variants will be even smaller and will have little or no impact on the familial risk of depression.12, 13, 14 The proportion of variance of depression risk explained by common genetic variants was estimated at 21%,15, 16 suggesting that there is influence of additional mechanisms such as rare genetic variants, which have not yet been as rigorously studied for depression as the common genetic variants. We have recently identified a rare missense variant in the LIPG17 gene and several coding variants in NKPD118 gene associated with depressive symptoms. The estimated impact of these variants on depressive symptomology is large.17, 18 In addition, two relatively small studies have nominally implicated rare genetic variants in ZNF34 and Cav2-adaptor genes in depression.19, 20 These studies corroborate the contribution of rare variants to the etiology of depression.
Next-generation sequencing (NGS) has greatly facilitated the opportunity to study rare variation in the human genome.21 However, as statistical power in a genetic association study is partly determined by the frequency of the genetic variant, large sample sizes are required to have sufficient statistical power to implicate rare variants with high confidence.22 As NGS is still expensive, large-scale whole-genome/exome sequencing is not yet feasible,21 which impedes the discovery of rare genetic variants through GWAS-like studies. However, unlike common variants (with a few exceptions), rare variants may also be studied in small samples in family-based studies.21 As both alleles from the parent are equally likely to be transmitted to the offspring, rare alleles can be enriched in families and can be localized through positional cloning.23, 24, 25 Further, individuals from a family are likely to share the same phenotype and environmental factors26 besides genetics.
Apart from family-based studies other designs including extreme-phenotype sampling22 and use of population isolates have been suggested to increase the chances of the discovery of rare genetic variants21 in smaller samples. Although individuals with extreme phenotypes are expected to be more homogenous and enriched for rare variants with large effects,25 isolated populations, besides being characterized by reduced phenotypic and genotypic heterogeneity, are also enriched for rare alleles due to isolation and genetic drift.27 For a complex and heterogeneous trait like depression, these factors can help increase the power to detect genetic variation that is rare in the general population. There is, however, a risk with such designs that the identified variants may not be relevant for the general population.21, 22
In the current study, to identify rare genetic variants associated with depression, we performed positional cloning using sparse genotyping in individuals selected for a high burden of depressive symptoms and clustered in families from an isolated population. Next, we identified the relevant mutations using NGS in informative individuals and families. We then replicated our findings in a cosmopolitan population. Finally, to understand the mechanisms through which the identified gene may be relevant for depression, we performed gene expression analysis in human and mice brain to localize the cell types in which the gene harboring the identified mutation is expressed.
Material and Methods
Study populations
The discovery population consisted of subjects from the Erasmus Rucphen Family (ERF), which is a genetically isolated community in the South-West of the Netherlands, and studied as part of the Genetic Research in Isolated Populations (GRIP) program.28 ERF includes ~3000 individuals who are living descendants of 22 founder couples who each had at least six children baptized in the community church. All data were collected between 2002 and 2005. The population exhibits minimal immigration and high inbreeding, resulting in a skewed distribution of rare allele frequencies.28 Depressive symptoms were evaluated using the Center for Epidemiologic Studies Depression (CES-D) and the Hospital Anxiety and Depression Scale (HADS). The HADS is a 14-item scale of which 7 relate to anxiety and 7 to depression. For depressive symptoms (HADS-D), the total score ranges from 0 to 21 with a higher score representing a higher burden of depressive symptoms. A cutoff of 11 has been validated as indicative of a current major depressive episode.29, 30
The replication cohort included subjects from the Rotterdam study (RS). RS is a prospective, population-based study from the district of Ommoord within the city of Rotterdam, designed to investigate the occurrence and determinants of diseases in the elderly.31 The cohort was initially defined in 1990 among 7983 persons who underwent a home interview and extensive physical examination at baseline and follow-up examinations occurring every 3–4 years (RS-I). The cohort was further extended in 2000 (RS-II) and 2005 (RS-III), establishing a total of 14 926 participants.31 RS is an outbred population, predominantly of Dutch origin. Participants from RS-I were assessed during four follow-up visits for depressive symptoms using the 20-item version of the CES-D scale. Data from the third follow-up visit was used in this study to maximize the sample size.
This study was approved by the Medical Ethical Committee of the Erasmus University Medical Center (Rotterdam, The Netherlands). All participants provided written informed consent to participate.
Genotyping
Linkage array
Genotyping was performed on 6 K Illumina Linkage IV Panels (Illumina, San Diego, CA, USA). After removing variants with a low call rate (<98%) or diverging from Hardy–Weinberg equilibrium (P-value<10−08), and removing individuals with a low call rate (<96%), 5250 single-nucleotide polymorphisms were retrieved from 3018 individuals. Mendelian inconsistencies were designated as missing.
Exome sequencing/genotyping
The exomes of 1336 individuals from the ERF study were sequenced at the Center for Biomics (Department of Cell Biology, Erasmus MC, Rotterdam, The Netherlands). Sequencing was performed at a mean depth of 74x (Agilent Technologies, Santa Clara, CA, USA, version 4 capture) on an Illumina Hiseq2000 sequencer using the TruSeq Version 3 protocol. After quality control we retrieved 543 954 high-quality single nucleotide variants (SNVs) in 1327 individuals. Individuals, whose exomes were not sequenced (N=1527) were genotyped on the Illumina Infinium HumanExome BeadChip, version 1.1. Calling was performed with GenomeStudio. In RS, exomes of 2628 randomly selected individuals from the RS-I population were sequenced at an average depth of 54 × (Nimblegen SeqCap EZ V2 capture) on an Illumina Hiseq2000 sequencer using the TrueSeq Version 3 protocol. The final dataset after quality control consisted of 600 806 SNVs among 2356 individuals, of which 1604 had CES-D data available. Details of sequencing and genotyping are provided in the supplement.
Statistical analyses
Linkage and haplotype analyses
To identify loci that might harbor large-effect-rare variation, we performed an affected-only linkage analysis using individuals from ERF scoring >90th percentile (score >12) of the HADS-D distribution (N=218; Supplementary Table 1). These 218 affected individuals were related to each other in >10 generations in a large multiplex pedigree consisting of 6562 individuals. PEDCUT software32 was used to cluster the 218 affected individuals into 48 non-overlapping sub-pedigrees to facilitate linkage analysis. Multipoint non-parametric and parametric linkage analyses were performed using MERLIN 1.1.2.33 Parametric linkage analysis was performed assuming incomplete penetrance and no phenocopies under dominant (0, 0.5, 0.5) and recessive models (0, 0, 0.5).34 Subsequently, to identify sub-families contributing to each linkage peak (henceforth referred to as contributing families), a ‘per family’ linkage analysis was performed for each locus of interest. To identify individuals contributing to the linkage scores of each locus, we constructed haplotypes using SIMWALK2.35 Linked regions for which we could ascertain a distinct segregating haplotype were followed up with fine mapping.
Identification of variants under the linkage peaks
Exome sequence data was used to identify variants that could explain the observed linkage peaks in the ERF discovery cohort. For each linkage region, we considered only rare (minor allele frequency (MAF)<0.05) and coding (missense, stop-coding) SNVs located within the corresponding haplotypes. To identify variants that could explain the corresponding linkage signal, we searched for variants shared by at least two affected haplotype carriers. Candidate variants were subjected to quantitative trait association analysis with depressive symptoms adjusted for age and gender using the ‘fastAssoc’ option in MERLIN 1.1.2. In RS, the replication cohort, quantitative trait association analysis of depressive symptoms (CES-D-scale) adjusted for age and gender was performed using seqMeta (v1.4) (http://cran.r-project.org/web/packages/seqMeta/index.html).
Human brain material
All procedures with human tissue were performed with the approval of the Medical Ethical Committee of the Erasmus MC Rotterdam, including written consent of all subjects for brain donation in accordance with Dutch license procedures and the Declaration of Helsinki. Fresh-frozen postmortem tissue blocks containing the middle frontal gyrus (BA9) from three donors were obtained from the Erasmus MC Department of Pathology (Supplementary Table 2). Donors were confirmed to have no past medical history of any known psychiatric or neurologic illness, with additional confirmation of the absence of clinical neuropathology by autopsy examination.
Human brain immunohistochemistry
Middle frontal gyrus tissue blocks were postfixed for 7 days in 4% paraformaldehyde (0.1 M phosphate buffer, pH 7.3) at 4 °C. Tissue was subsequently transferred to 10% sucrose (0.1 m phosphate buffer, pH 7.3) and stored overnight at 4 °C. Embedding was performed in 12% gelatin/10% sucrose, with fixation in 10% paraformaldehyde/30% sucrose solution for 4 h at room temperature and overnight immersion in 30% sucrose at 4 °C. Serial 40 μm sections were collected along the rostrocaudal axis using a freezing microtome (Leica, Wetzlar, Germany; SM 2000 R) and stored at −20 °C in a solution containing 37.5% ethylene glycol (Avantor, Central Valley, PA, USA, 9300), 37.5% glycerol (VWR Chemicals, Radnor, PA, USA, 24 386.298) and 25% 0.1 m phosphate buffer. Free-floating sections were washed thoroughly with PBS before being incubated in sodium citrate (10 mm) at 80 °C for 45 min and rinsed with PBS. Sections were pre-incubated with a blocking PBS buffer containing 1% Triton X-100 and 5% bovine serum albumin for 1 h at room temperature.
Primary antibody labeling was performed in PBS buffer containing 1% Triton X-100 and 1% BSA for 72 h at 4 °C. The following primary antibodies were used: Rabbit anti-RCL1 (1:250, Sigma, St Louis, MO, USA, SAB4500053), Mouse anti-GFAP (1:2000, Sigma, G6171), Mouse anti-MAP2 (1:200, EMD Millipore, Merck Life Sciences, Darmstadt, Germany, MAB3418), Guinea pig anti-MAP2 (1:1000, Synaptic Systems, Goettingen, Germany, 188 004), Mouse anti-SMI31 (1:250, Affinity, Exeter, UK, NA1219), Rat anti-CD44 (1:400, Sigma, SAB4700188). Following primary antibody labeling, sections were washed with PBS and then incubated with corresponding Alexa-conjugated secondary antibodies and cyanine dyes (1:400, Braunschweig Chemicals, Amsterdam, The Netherlands) in PBS buffer containing 1% Triton X-100, 1% BSA for 4 h at room temperature. Nuclear staining was performed using DAPI (1:10 000, Thermo Fisher Scientific, Waltham, MA, USA). Images were acquired using a Zeiss LSM 700 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Mouse brain immunohistochemistry
All mouse experiments were approved by the local animal welfare committee. Three adult C57BL/6J mice were killed by standard transcardiac perfusion with saline followed by 4% paraformaldehyde. Brains were dissected and postfixed in 4% paraformaldehyde for 2 h at room temperature. Subsequently, 40 μm mouse brain sections were prepared and stained with the same procedures as the human brain sections, with the exceptions that they were: (a) not incubated in sodium citrate, (b) incubated in 0.5% Triton X-100 instead of 1%, (c) blocked with horse serum (Invitrogen) instead of BSA and (d) Mouse anti-GFAP (Sigma, G6171) was used in a 1:20 000 dilution.
Results
Linkage and haplotyping
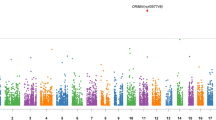
Results of genome-wide linkage analysis are provided in Supplementary Figure 1. There were 10 genomic regions including 5q14, 9p22-24, 10q21, 11q13, 12q24, 15q21, 19q13, 20p13, 21q22 and 21q24 that showed a log of odds score >3 (Table 1). Chromosome 9p22-24 showed over 11 mega-bases (Mb) long haplotype segregating in three generations of the contributing family. The haplotype was shared by all of the affected individuals (N=8) in this family (Supplementary Figure 2) including two homozygous carriers. For linked regions including chromosomes 15q21, 19q13, 20p13 and 21q22, we observed small (<190 kilo-base (kb)) haplotypes with mixed segregation patterns (Supplementary Figures 3–6). For chromosomes 5q14, 10q21 and 11q13 we could not identify clear segregating haplotypes.
Fine mapping using exome sequencing
Upon filtering the exome sequence data based on sharing among affected haplotype carriers, we identified a rare c.1114C>T missense variant (rs115482041) in 6 of the 8 affected haplotype carriers in the 9p22-24 region in the RCL1 gene (Supplementary Table 3; Supplementary Figure 7). Within the remaining linkage regions, including 15q21, 19q13, 20p13 and 21q22, we searched for shared variants in heterozygous, homozygous and compound heterozygous forms but identified none that could explain linkage signals in these regions. Rs115482041 was sequenced with a high confidence (read depth=39 × ) and showed a base call accuracy of >99.99% (Phred scale quality=94). The two homozygous carriers of the haplotype (brothers) were also homozygous for rs115482041-T (Supplementary Figure 7). These two siblings were diagnosed with major depressive disorder (MDD), whereas their sister and her son, who were heterozygous for the rare variant, were both diagnosed with depressive disorder not otherwise specified. The MAF of rs115482041 was 1% in the ERF study compared to 0.16% in 1000 Genomes (http://www.1000genomes.org/) and 0.33% in the Exome Sequencing Project (ESP; http://evs.gs.washington.edu/EVS/). Rs115482041 is a conserved protein coding variant (genomic evolutionary rate profiling=4.0) and predicted to affect protein function (p.L186F) by three different algorithms (PolyPhen2, 0.68; SIFT, 0.01; MutationTaster, 0.99; combined annotation dependent depletion score, 12.15).
As ERF is a large pedigree spread over 23 generations, we extended this sub-family to more distant relatives to identify other potential carriers to confirm the segregation of rs115482041 (Supplementary Figure 8). We performed Sanger sequencing of 30 individuals with available HADS-D score from the extended family including two haplotype carriers that did not carry the rs115482041_T variant based on NGS. We identified three more heterozygous carriers of rs115482041_T, two of whom had a high HADS depression score (Supplementary Figure 8). In total, there were 54/2816 rs115482041_T carriers who were related to each other across five generations (Figure 1). No Mendelian inconsistencies were observed. A significant association of rs115482041 with the HADS-D was observed (N=2393, log of odds=3.24, P-value=1x10−4). The effect of the rare allele (T) on HADS-D was large (β=2.33), indicating a mean increase of 2.33 points for each mutant allele (T) (2.33 points for heterozygote C/T carriers and 4.66 points for homozygous T/T carriers, compared with the reference C/C carriers). The heritability of the HADS-D in the extended sub-pedigree (Supplementary Figure 8) was estimated to be 94%, and 54% of the variance in depressive symptoms was explained by c.1114C>T. Across the entire ERF cohort, the heritability of the HADS-D was estimated to be 22%, of which rs115482041 explained 0.64% of the variance in depressive symptoms (2.9% of the genetic variance of HADS-D). We found no evidence to suggest that this association was due to an underlying somatic condition, including cancer, cardiovascular, lung, liver or metabolic phenotypes.

All carriers of c.1114C>T in the Erasmus Rucphen Family (ERF) study. Males are depicted with squares and females with circles. The text below each individual shows his/her age followed by the score on HADS-D scale, genotype and psychiatric diagnosis (mdd refers to major depressive disorder/lifetime depression and mild refers to depression not otherwise specified), if any. Individuals connected with dotted lines are duplicates. Dots indicate missing values. HADS, Hospital Anxiety Depression Scale.
For 17 of the 54 carriers, we were able to obtain medication prescriptions at the time of the HADS assessments. Six were using antidepressant medication and/or benzodiazepines at the time of assessment including selective serotonin reuptake inhibitors (paroxetine, citalopram), non-selective serotonin reuptake inhibitors (imipramine, amitriptyline) and other antidepressants (mirtazapine) coupled with benzodiazepine derivatives (temazepam, oxazepam, alprazolam and bromazepam).
In the replication cohort, we identified 23 rs115482041_T carriers (of whom 17 had completed the CES-D) from a total of 2356 individuals (MAF=0.5%). The rs115482041 variant was sequenced at an average depth of 39 × and showed a base call accuracy of >99.99% (Phred scale quality=70). A significant association with CES-D was observed (P-value=0.03). The effect of rs115482041_T on the CES-D scale was large in RS (β=3.60, δβ=1.69), which was comparable to that in ERF (CES-D; β=3.47, δβ=1.74).
RCL1 is abundantly expressed in interlaminar astrocytes in human cerebral cortex
RCL1 is an RNA 3′-terminal phosphate cyclase-like protein that is highly conserved across eukaryotic species.36 The function of the RCL1 protein has predominantly been studied in yeast, where it is has been shown to have nuclease activity that separates 18 S transcripts from the precursor rRNA transcript by co-transcriptional endonucleolytic cleavage. In line with this function, RCL1 has been shown in yeast to have a nucleolar subcellular localization.37
No previous study has investigated the cell-type specific expression of RCL1 in human brain. Immunohistochemistry of human postmortem cerebral cortex revealed nuclear labeling of RCL1 in most cells (Supplementary Figure 9a), including neurons (Supplementary Figure 9b). In addition, a distinct pattern of long unbranched tortuous processes often extending from layer 1 to layer 3/4 exhibited abundant RCL1 expression (Figure 2). These processes showed no co-localization with markers of neuronal processes including MAP2 (dendritic, Supplementary Figure 10a) or SMI31 (axonal, Supplementary Figure 10b). In contrast, strong co-localization was observed with the astrocyte markers GFAP and CD44 (Figure 2; Supplementary Figure 11) with their cell bodies located in layer 1 of the cortex and a cellular morphology consistent with primate-specific interlaminar astrocytes that extend very long tortuous processes often extending more than a millimeter in length into cortical layers 2–4.38 The specificity of RCL1 expression in layer 1 interlaminar astrocytes was additionally highlighted by the absence of cytoplasmic RCL1 labeling in GFAP+ astrocytes in layer 5/6 of the cerebral cortex (Supplementary Figure 12). Moreover, consistent with the recent evolution of interlaminar astrocytes limited to primates, we observed no significant cytoplasmic RCL1 labeling in GFAP+ astrocytes of the mouse cerebral cortex (Supplementary Figure 13). Immunohistochemistry controls for RCL1 labeling, autofluorescence and nonspecific secondary antibody labeling are presented in Supplementary Figures 14 and 15.

Immunohistochemical labeling of RCL1 in human cerebral cortex. (a) Overview of RCL1 labeling showing co-localization with long (>350 μm) tortuous interlaminar GFAP-positive extensions (the dashed line marks the approximate border between cortical layers I and II, scale bar=50 μm). (b) Higher magnification of marked region in a (scale bar=12 μm).
Discussion
Using a three-stage design in a family-based setting to discover rare genetic variants influencing depression, we identified a rare genetic variant (rs115482041) on chromosome 9p24 in the RCL1 gene that segregated with depression across multiple generations in an extended family. The variant was estimated to explain more than half of the variation in depressive symptoms in the extended family, and 2.9% of the heritability in the overall genetically isolated ERF population, which is enriched for rs115482041. The strong association with depressive symptoms was further replicated in the population-based RS cohort.
Rs115482041 is a coding variant (c.1114C>T, p.L186F) located in a highly conserved RNA Terminal Phosphate Cyclase-Like 1 domain and predicted as damaging. The variant exists only in 8/5008 alleles (MAF=0.16%) in 1000 Genomes and 43/12963 alleles (MAF=0.33%) in the Exome Variant Server. It occurs more frequently in European-Americans (37/8563, MAF=0.43%) compared with the African-Americans (6/4400, MAF=0.14%). In contrast, the ERF pedigree is considerably enriched for rs115482041 carriers (56/5632 alleles, MAF=1%). As a consequence, we had considerably increased power to detect the association with depression. Further, of interest is that both of the homozygous carriers from ERF exhibited high scores on the depressive symptom rating scales and were also diagnosed with MDD requiring a combination of psychotherapy and antidepressant treatment. The association of rs115482041 with depressive symptoms in the population-based Rotterdam Study further confirms the robustness of the finding and its relevance beyond the ERF cohort. We could not find our variant in the top 10 000 findings in the GWAS performed by 23andMe.11 As the variant rs115482041 is extremely rare in 1000 Genomes, it is very likely that the variant may not have been analyzed because of poor imputation quality39 (in ERF the imputation quality of rs115482041 based on the same release of 1000 Genomes is 0.09).
RCL1 is ubiquitously expressed in eukaryotes with a demonstrated function in ribosome biogenesis. Our findings now suggest that RCL1 might have additional functions in the human brain. In contrast to the mouse brain, immunohistochemistry of RCL1 expression in human brain identified abundant labeling of interlaminar astrocytes in layer 1 of the cerebral cortex, with their characteristic tortuous processes extending long distances of hundreds of microns into deeper cortical layers. Notably, recent studies in rodent models40, 41 and humans42, 43, 44 have proposed novel pathophysiological functions of astrocytes in MDD. However, the neurobiological function of interlaminar astrocytes remains largely unknown. Interestingly, their extensive projections have been hypothesized to form a network for long-range coordination of intra-cortical communication.38, 45, 46, 47 On the other hand, we also found evidence for the presence of RCL1 in neurons for which we cannot yet distinguish the relative contribution of neuronal and/or astrocyte dysfunction that might explain the genetic association with RCL1 c.1114C>T (p.L186F). Future studies should be performed to elucidate the function of interlaminar astrocytes and their potential influence on the pathophysiology of depression.
In this study we used a unique multistep approach to identify rare genetic variants that confer large effects on depression. Our study design had several advantages. First, the discovery cohort ERF was enriched for the rs115482041_T variant, which led to an increase in statistical power to detect genotype/phenotype association. Secondly, using haplotype analysis we were able to identify the most relevant individuals for each locus that facilitated the identification of segregating candidate variants using the filtering/sharing approach. Finally, by combining linkage, haplotype sharing and filtering, we contained the problem of multiple testing. Importantly however, despite these distinct advantages we were able to identify a genetic variant for only one of the several linked genomic regions, for which, there may be several reasons including structural variants, and intronic or intergenic single-nucleotide variants that were not evaluated in the current study. Notably, many of the other haplotype regions were relatively small (a few kb) and therefore of questionable relevance, compared with that of the 9p22-24 locus which was nearly 11 Mb. As ERF is a young genetic isolate with high rate of consanguinity, 500 kb–1.5 Mb homozygous blocks may just be shared by chance.48 Notably, however, we previously found that some of these additional loci are significantly linked with personality traits, including conscientiousness (20p13), extraversion (12q24), openness (12q24 and 19q13), neuroticism (19q13 and 21q22-24) and agreeableness (21q22).49
In summary, our findings identify RCL1 as an important candidate gene for depression in a region that shows genome-wide significant linkage with depressive symptoms in the general population. We show that the RCL1 protein is present in human brain, including neurons and astrocytes. Further research is needed to elucidate the mechanism of RCL1 in the pathogenesis of depression.
References
Lohoff FW . Overview of the genetics of major depressive disorder. Curr Psychiatry Rep 2010; 12: 539–546.
Paykel ES, Brugha T, Fryers T . Size and burden of depressive disorders in Europe. Eur Neuropsychopharmacol 2005; 15: 411–423.
Ayuso-Mateos JL, Vazquez-Barquero JL, Dowrick C, Lehtinen V, Dalgard OS, Casey P et al. Depressive disorders in Europe: prevalence figures from the ODIN study. Br J Psychiatry 2001; 179: 308–316.
Jacobi F, Wittchen HU, Holting C, Hofler M, Pfister H, Muller N et al. Prevalence, co-morbidity and correlates of mental disorders in the general population: results from the German Health Interview and Examination Survey (GHS). Psychol Med 2004; 34: 597–611.
CONVERGE Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015; 523: 588–591.
Demirkan A, Lahti J, Direk N, Viktorin A, Lunetta KL, Terracciano A et al. Somatic, positive and negative domains of the Center for Epidemiological Studies Depression (CES-D) scale: a meta-analysis of genome-wide association studies. Psychol Med 2016; 46: 1613–1623.
Dunn EC, Brown RC, Dai Y, Rosand J, Nugent NR, Amstadter AB et al. Genetic determinants of depression: recent findings and future directions. Harv Rev Psychiatry 2015; 23: 1–18.
Hek K, Demirkan A, Lahti J, Terracciano A, Teumer A, Cornelis MC et al. A genome-wide association study of depressive symptoms. Biol Psychiatry 2013; 73: 667–678.
Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511.
Sullivan PF . Genetics of disease: associations with depression. Nature 2015; 523: 539–540.
Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016; 48: 1031–1036.
Gibson G . Rare and common variants: twenty arguments. Nat Rev Genet 2011; 13: 135–145.
Maher B . Personal genomes: The case of the missing heritability. Nature 2008; 456: 18–21.
Offit K . Personalized medicine: new genomics, old lessons. Hum Genet 2011; 130: 3–14.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 2013; 45: 984–994.
Levinson DF, Mostafavi S, Milaneschi Y, Rivera M, Ripke S, Wray NR et al. Genetic studies of major depressive disorder: why are there no genome-wide association study findings and what can we do about it? Biol Psychiatry 2014; 76: 510–512.
Amin N, Jovanova O, Adams HH, Dehghan A, Kavousi M, Vernooij MW et al. Exome-sequencing in a large population-based study reveals a rare Asn396Ser variant in the LIPG gene associated with depressive symptoms. Mol Psychiatry 2016; doi:10.1038/mp.2016.101; e-pub ahead of print 19 July 2016.
Amin N, Belonogova NM, Jovanova O, Brouwer RWW, Rooij JGJv, Hout MCGNvd et al. Non-synonymous variation in NKPD1 increases depressive symptoms in the European populations. Biol Psychiatry 2016; doi:10.1016/j.biopsych.2016.08.008; e-pub ahead of print.
Pirooznia M, Wang T, Avramopoulos D, Potash JB, Zandi PP, Goes FS . High-throughput sequencing of the synaptome in major depressive disorder. Mol Psychiatry 2016; 21: 650–655.
Subaran RL, Odgerel Z, Swaminathan R, Glatt CE, Weissman MM . Novel variants in ZNF34 and other brain-expressed transcription factors are shared among early-onset MDD relatives. Am J Med Genet B Neuropsychiatr Genet 2016; 171: 333–341.
Auer PL, Lettre G . Rare variant association studies: considerations, challenges and opportunities. Genome Med 2015; 7: 16.
Lee S, Abecasis GR, Boehnke M, Lin X . Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014; 95: 5–23.
Ott J, Wang J, Leal SM . Genetic linkage analysis in the age of whole-genome sequencing. Nat Rev Genet 2015; 16: 275–284.
Pulst SM . Genetic linkage analysis. Arch Neurol 1999; 56: 667–672.
Cirulli ET, Goldstein DB . Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 2010; 11: 415–425.
Ott J, Kamatani Y, Lathrop M . Family-based designs for genome-wide association studies. Nat Rev Genet 2011; 12: 465–474.
Hatzikotoulas K, Gilly A, Zeggini E . Using population isolates in genetic association studies. Brief Funct Genomics 2014; 13: 371–377.
Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS . The effect of genetic drift in a young genetically isolated population. Ann Hum Genet 2005; 69: 288–295.
Bjelland I, Dahl AA, Haug TT, Neckelmann D . The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res 2002; 52: 69–77.
Hung CI, Liu CY, Wang SJ, Yao YC, Yang CH . The cut-off points of the Depression and Somatic Symptoms Scale and the Hospital Anxiety and Depression Scale in detecting non-full remission and a current major depressive episode. Int J Psychiatry Clin Pract 2012; 16: 33–40.
Hofman A, Darwish Murad S, van Duijn CM, Franco OH, Goedegebure A, Ikram MA et al. The Rotterdam Study: 2014 objectives and design update. Eur J Epidemiol 2013; 28: 889–926.
Liu F, Kirichenko A, Axenovich TI, van Duijn CM, Aulchenko YS . An approach for cutting large and complex pedigrees for linkage analysis. Eur J Hum Genet 2008; 16: 854–860.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR . Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Durner M, Vieland VJ, Greenberg DA . Further evidence for the increased power of LOD scores compared with nonparametric methods. Am J Hum Genet 1999; 64: 281–289.
Sobel E, Lange K . Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 1996; 58: 1323–1337.
Horn DM, Mason SL, Karbstein K . Rcl1 protein, a novel nuclease for 18 S ribosomal RNA production. J Biol Chem 2011; 286: 34082–34087.
Billy E, Wegierski T, Nasr F, Filipowicz W . Rcl1p, the yeast protein similar to the RNA 3′-phosphate cyclase, associates with U3 snoRNP and is required for 18 S rRNA biogenesis. EMBO J 2000; 19: 2115–2126.
Oberheim NA, Wang X, Goldman S, Nedergaard M . Astrocytic complexity distinguishes the human brain. Trends Neurosci 2006; 29: 547–553.
Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat Commun 2015; 6: 8111.
Banasr M, Duman RS . Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry 2008; 64: 863–870.
Cao X, Li LP, Wang Q, Wu Q, Hu HH, Zhang M et al. Astrocyte-derived ATP modulates depressive-like behaviors. Nat Med 2013; 19: 773–777.
Nagy C, Suderman M, Yang J, Szyf M, Mechawar N, Ernst C et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol Psychiatry 2015; 20: 320–328.
Rajkowska G, Stockmeier CA . Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets 2013; 14: 1225–1236.
Rajkowska G, Hughes J, Stockmeier CA, Javier Miguel-Hidalgo J, Maciag D . Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol Psychiatry 2013; 73: 613–621.
Colombo JA, Sherwood CC, Hof PR . Interlaminar astroglial processes in the cerebral cortex of great apes. Anat Embryol 2004; 208: 215–218.
Colombo JA, Reisin HD . Interlaminar astroglia of the cerebral cortex: a marker of the primate brain. Brain Res 2004; 1006: 126–131.
Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F et al. Uniquely hominid features of adult human astrocytes. J Neurosci 2009; 29: 3276–3287.
McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L et al. Runs of homozygosity in European populations. Am J Hum Genet 2008; 83: 359–372.
Amin N, Schuur M, Gusareva ES, Isaacs A, Aulchenko YS, Kirichenko AV et al. A genome-wide linkage study of individuals with high scores on NEO personalitytraits. Mol Psychiatry 2012; 17: 1031–1041.
Acknowledgements
Erasmus Rucphen Family Study: The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP Grant Number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/Grant Agreement HEALTH-F4-2007-201413 by the European Commission under the programme ‘Quality of Life and Management of the Living Resources’ of 5th Framework Programme (No. QLG2-CT-2002-01254) and coSTREAM project number 105591. High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). Exome-sequencing analysis in ERF was supported by the ZonMw grant (Project 91111025). We are grateful to all study participants and their relatives, general practitioners and neurologists for their contributions and to P Veraart for her help in genealogy, J Vergeer for the supervision of the laboratory work and P Snijders for his help in data collection. Najaf Amin and Femke MS de Vrij are supported by the Hersenstichting Nederland (the Netherlands Brain Foundation) (Project Numbers F2013(1)-28 and F2012(1)-39).
Rotterdam study: The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII) and the Municipality of Rotterdam. We are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists.
The generation and management of the exome sequencing data for the Rotterdam Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Netherlands. The Exome-Sequencing dataset was funded by the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) sponsored Netherlands Consortium for Healthy Aging (NCHA; Project No. 050-060-810), by the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by the and by a Complementation Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL; www.bbmri.nl; Project Number CP2010-41). We thank Mr Pascal Arp, Ms Mila Jhamai and Mr Marijn Verkerk for their help in creating the RS-Exome-Sequencing database.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website .
Supplementary information
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Amin, N., de Vrij, F., Baghdadi, M. et al. A rare missense variant in RCL1 segregates with depression in extended families. Mol Psychiatry 23, 1120–1126 (2018). https://doi.org/10.1038/mp.2017.49
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.49
This article is cited by
-
Integration of whole-exome sequencing and structural neuroimaging analysis in major depressive disorder: a joint study
Translational Psychiatry (2024)
-
Contemporary Genome-Wide Association Studies in Depression: The Critical Role of Phenotyping
Neuroscience and Behavioral Physiology (2022)
-
Modification by genetic polymorphism of lead-induced IQ alteration: a systematic review
Environmental Science and Pollution Research (2022)
-
RCL1 copy number variants are associated with a range of neuropsychiatric phenotypes
Molecular Psychiatry (2021)
-
Looking into the genetic bases of OCD dimensions: a pilot genome-wide association study
Translational Psychiatry (2020)
