
Abstract
Historically, only cells of the adaptive immune system have been considered capable of retaining memory for infectious challenges. Recently, however, cells of the innate immune system have been shown to be capable of displaying long-term functional memory following a single immunostimulatory challenge, leading to enhanced production of proinflammatory molecules upon other subsequent, and temporally distant, immunostimulatory challenges. This effect has been termed ‘trained innate immunity’, and is underwritten by stable epigenetic changes in immune and metabolic pathways. Importantly, the long-term training of innate immune cells can occur as a result of infectious as well as and non-infectious challenges, including stress. Given the role that both stress and an activated immune system have in neuropathology, innate immune training has important implications for our understanding and treatment of neuropsychiatric disorders. This review focuses on the evidence for trained innate immunity and highlights some insights into its relevance for psychiatric diseases.
Similar content being viewed by others

Introduction
(Neuro)inflammation is increasingly recognized as a critical factor in the pathogenesis of numerous psychiatric illnesses. Indeed, research on immune dysregulation and psychiatric disorders has gained significant attention in recent years as data gathered from numerous experimental models have converged to provide support for the role of inflammatory mechanisms in the etiology of depression, schizophrenia and related psychotic disorders.1 Over the last 5 years, the number of publications on immunity and depression alone has almost doubled.2 Similarly, in the past 7 years, data supporting an immune-mediated cause in schizophrenia have also increased substantially.3 This advocates that research is now entering a new era of immunopsychiatric investigations that will profoundly change the understanding of brain disorders, in which immune impairments will be now acknowledged as potential underlying mechanisms of altered behavioral manifestations.
Increasingly, evidence proposes that proinflammatory molecules directly exert negative effects on neuronal cells, ultimately contributing to the development and progression of major depressive disorder, schizophrenia and post-traumatic stress disorder, amongst others.4, 5 Cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor-α (TNF-α), interferon-α (IFN-α) and interferon-γ (IFN-γ), as well as reactive oxygen species and reactive nitrogen species, cause disruptions in neurogenesis, neuronal excitability, synaptic transmission, synaptic plasticity and neuronal survival.6, 7 These disturbances have been implicated in the development and progression of neuropsychiatric disorders.8 Microglia, the resident innate immune cells of the central nervous system, are the major mediators of neuroinflammation,9 although monocytes and macrophages are also important innate immune sources of sources of proinflammatory molecules that can act in the central nervous system.10, 11 These cells are strongly implicated in the pathogenesis of neuropsychiatric disorders therefore.12–14
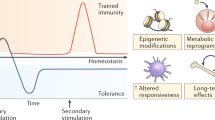
Over the past decades, the major difference between the adaptive and innate immune systems has been considered the ability of the adaptive immune system to retain memory for specific infectious agents, whilst the innate immune system has been considered neither capable of memory nor specificity.15 This memory function of the adaptive immune system is critical to immunity in vertebrates, often offering lifelong protection against re-infection. Recently, however, cells of the innate immune system, including monocytes and macrophages, have been discovered capable of long-term functional memory following a single short-lived immunostimulatory event, resulting in markedly enhanced production of proinflammatory molecules in response to a subsequent, temporally distant, immunostimulatory event. This has been termed ‘trained innate immunity’ or ‘innate immune memory’.16, 17 In animal models of dysfunctional or absent adaptive immunity, trained innate immune cells can protect against lethal infection with various pathogens.18, 19 Trained innate immunity represents a paradigm shift therefore in our understanding of innate immunity, with important implications for disease pathogenesis and immunotherapeutics.16, 20
Trained innate immunity is distinct from priming. Priming is a well-described phenomenon during certain infections or stimulations.16, 20, 21 Often, pre-exposure of an innate immune cell to a cytokine such as IFN-γ is necessary for the cell to be able to fully activate and produce a robust immune response when exposed to a pathogen.22, 23 The cells are said to be pre-activated or ‘primed’ in response to stimulation with the cytokine. Typically, primed cells produce little, if any, cytokines in the absence of a contemporaneous co-stimulation by a pathogen.24, 25 Priming effects also rapidly decline. In contrast, training is typically induced by pathogen-associated molecular patterns (PAMPs), and involves long-term changes in innate immune cell function that persist well beyond the primary immunostimulatory event. Unlike primed cells, trained cells often have an elevated baseline proinflammatory cytokine production. The mechanisms underlying training are distinct from priming. Priming often involves the mobilization of pre-formed receptors in the cytoplasm to the plasma membrane as a result of a stimulus, leading to short-term term changes in receptor number or affinity.16, 20 In contrast, training is underwritten by stable epigenetic changes in immune and metabolic pathways.16, 20, 21 This long-term memory effect can persist for months, and possibly years, and is not exclusive or specific to the type of primary immune-stimulating event.26 Recent studies suggest that innate immune cells also retain long-term memory for non-infectious immunostimulatory challenges, such as stress.27
Despite the widespread interest in immune function and psychiatric illness, there has been no exhaustive discussion relating to the implications of trained innate immunity on mental health pathology. Considering that trained innate immunity represents a paradigm shift in our understanding of immune function,28 the theoretical contributions of trained innate immunity to illnesses such as depression are potentially marked and worth of a comprehensive analysis. Here we review recent evidence for innate immune training and examine why it is likely to be a major contributing factor to the pathogenesis of neuropsychiatric disorders. We concentrate primarily on monocytes, macrophages and microglia, though there is also extensive evidence for trained immunity in natural killer cells,16 which have themselves been recently hypothesized to have a role in psychiatric illness.29
Trained innate immunity
The innate immune system and training
The innate immune system recognizes invading pathogens through receptors called pathogen recognition receptors (PRRs), which are present on the surface, in the endosome or in the cytoplasm of innate immune cells.30 PRRs recognize conserved microbial molecules, including lipids, proteins, nucleic acids and carbohydrates. These microbial molecules are known as PAMPs.30 A diverse range of PRR types exist, including toll-like receptors (TLRs), C-type lectin receptors, nucleotide-binding oligomerization domain-like receptors (NODs) and retinoic acid-inducible gene I helicases.30 A specific PRR may be capable of recognizing a single or multiple different PAMP. TLR-4, for example, recognizes lipopolysaccharide (LPS), an endotoxin present in the cell wall of multiple different gram-negative bacteria.31 Often, microorganisms will have multiple PAMPs, which will together stimulate a suite of PRR on host cells. Herpes simplex virus PAMP activate TLR-2 on the cell surface and TLR9 in the endosome,30 for example.
Typically, stimulation of a PRR by a PAMP leads to immune activation of the cell and the release of various proinflammatory cytokines, chemokines, reactive oxygen species and reactive nitrogen species.30 The exact response may vary dependent on the type of PAMP–PRR interaction. However, many different PAMP–PRR interactions activate the same or similar downstream signaling pathways. Activation of a PRR by a PAMP can sometimes lead to an altered cellular immune response upon subsequent reactivation of the same PRR or the activation of a different PRR that feeds into similar signaling pathways. This has been known about for some time in the context of innate immune tolerance.32, 33 Innate immune tolerance was first described in relation to the immunoparalysis that occurs after Gram-negative sepsis in critically ill patients.32 Here, cells of the innate immune system fail to mount an appropriate proinflammatory response to re-stimulation by LPS or Gram-negative bacteria following Gram-negative sepsis. Thus, innate immune cells appear to retain an inhibitory memory for high-dose LPS stimulation under certain conditions. Innate immune cells have also recently been shown to be capable of retaining a long-term memory for certain PAMP or pathogens, resulting in an exaggerated immune response upon subsequent re-stimulation with similar or dissimilar PAMP or pathogens. This heterogeneous proinflammatory memory effect, ‘trained innate immunity’, appears to be prolonged, lasting sometimes for months, and possibly even years.16 Whether tolerance or training occurs is dependent on a number of factors, including the amount, nature and duration of exposure of the PAMP. For example, whilst high-dose LPS results in decreased production of proinflammatory molecules upon re-exposure to LPS, low-dose LPS exposure results in increase production of proinflammatory molecules upon re-exposure to LPS.34
That this memory effect is heterogeneous and long lasting has important implications, as it may set the stage for a proinflammatory responsive state in response to a diverse array of temporally distant immunostimulatory events. Table 1 lists some of the main features of trained innate immunity, as well as potential consequences for neuropsychiatric disorders.
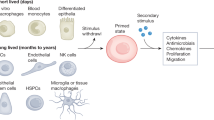
Training of monocytes and macrophages
Given that the adaptive immune system is highly specific, and the long-held view that the innate immune system displays no memory for pathogens, we would not expect an enhanced immune response upon re-infection at a distant time with dissimilar organisms. Nor would we expect an enhanced immune response upon re-stimulation in the absence of a functional adaptive immune system. Several studies performed decades ago demonstrated just this though. Mice immunized with Bacillus Calmette–Guérin (BCG) displayed enhanced immunity and protection against lethal infection with non-tuberculous organisms such as Candida albicans and Schistosoma mansoni.18, 35 T-lymphocyte depletion failed to dampen this protective effect, which was subsequently found to be mediated by activated tissue macrophages.18 Mice previously infected with the fungus C. albicans also showed enhanced immune responses and increased survival in response to infection with the Gram-positive bacterium Staphylococcus aureus,36 an effect that was again found to be dependent on macrophage responses.19
The importance of these findings and their implications were largely ignored however, until two recent studies replicated and extend these findings in human innate immune cells. Kleinnijenhuis et al. isolated monocytes from adult humans before and 2 weeks after BCG vaccination, and stimulated them ex vivo with BCG, S. aureus, C. albicans or LPS.26 Post BCG vaccination, monocytes showed markedly enhanced production (up to sevenfold) of various proinflammatory cytokines, including IFN-γ, TNF-α and IL-1β upon homogenous and heterogeneous stimulation, relative to monocytes pre-BCG vaccination. This upregulation in proinflammatory function was accompanied by increased expression of various activation markers and PRRs, including CD11b, CD14 and TLR-4, and was still present 1 year after BCG vaccination.26 Primary stimulation of NOD2, a PRR for mycobacteria, with the NOD2-specific ligand muramyl dipeptide, mimicked the training effects of BCG, which was prevented when inhibiting NOD2 during primary stimulation, as well as its downstream signaling pathway, Rip2 kinase. Such training effects were also seen in human monocytes following neonatal BCG vaccination.37
Similar training effects were observed following stimulation of adult human monocytes in vitro with either C. albicans or β-glucan (a component of fungal cell walls) for 24 h.38 Upon a second stimulation with C. albicans, bacteria or various PAMP/PRR ligands up to 2 weeks later, pre-stimulated monocytes showed significant increases (of up to 10-fold) in TNF-α and IL-6 production relative to naive monocytes. The magnitude of the training effects was dependent on the doses of both the primary and secondary stimulation. Again, specifically inhibiting the PRR for β-glucan and its downstream signaling pathways during primary stimulation, in this case Dectin-1 and Raf-1, prevented training. These training effects were accompanied by marked and long-lasting changes in gene expression. Genes for multiple PRR unrelated to Candida/β-glucan were upregulated, including several TLRs, which might partly explain the heterogeneous effect of training. Other upregulated genes included chemokines and nitric oxide synthases, as well as several histone methyltransferases, suggesting a potential role for epigenetic remodeling in training.
For some PAMP, the dose can determine whether training or tolerance is induced, whereas for others, different doses appear to consistently induce training. For example, moderate to high doses of a variety of PAMP and PRR ligands, such as LPS (100 μg ml−1), Pam3CSK4 (100 μg ml−1), flagellin (10 μg ml−1) and Poly(I:C) (100 μg ml−1) (which all engage TLRs) induced heterogeneous tolerance in human monocytes.34 Lower doses (0.1 pg ml−1 to 1 μg ml−1) however, induced heterogeneous training. In contrast, low to moderate doses of β-glucan (1 μg ml−1), Tri-DAP (10 μg ml−1) and muramyl dipeptide (10 μg ml−1) (which engage NODs), all induced training (up to fivefold increases in TNF-α). The reason why some PAMP/PRR induce only training or tolerance across a range of doses, whilst for other PAMP/PRR the effect is dose-dependent, is currently unclear.
Training of microglia
Few studies have investigated the long-term upregulation of microglial function as a result of a single immunostimulatory challenge, and none in the context of the concept of training. Nevertheless, there are suggestions that microglia are capable of being trained. Hippocampal microglia isolated from adult mice that had been injected once intraperitonealy with Salmonella typhimurium and then given an intra-hippocampal injection of LPS 4 weeks later showed an increase in the activation markers CD11c and MHCII relative to microglia from mice only exposed to S. typhimurium.39 S. typhimurium injection alone did not result in an increase in CD11c and MHCII immunoreactivity, suggesting that the increase seen following LPS injection was not simply due to an increase in the number of microglia. Neonatal rats injected with Escherichia coli subcutaneously and then given a peripheral injection of LPS in adulthood had increased microglial CD11b gene expression, as well as faster and more prolonged increases in microglial production of IL-1β protein, without there being any differences in microglial numbers.40, 41 These changes were accompanied by decreased neurogenesis in the hippocampus and memory impairments, which were prevented when caspase-1 (a protease that cleaves the precursors pro-IL-1β and pro-IL-18 into active IL-1β and IL-18) was inhibited before LPS challenge in adulthood. Although suggestive of training, it is possible, however, that interactions between the adaptive and the innate immune system were responsible for the observed effects in these studies. To specifically rule out interactions with other cell types, live microglia need to be isolated and tested in vitro. Although animals with severe combined immunodeficiency could be used to rule out interactions with the adaptive immune system, this would still leave the possibility that other cell types such as astrocytes and neurons, which are known to have immunomodulatory properties,42, 43 are responsible for any observed effects. A very limited number of studies have performed experiments on isolated microglia. Microglia isolated from adult rats that had been infected neonatally with E. coli showed increased production of IL-1β mRNA (on a per cell basis) when exposed to low-dose LPS in vitro.44 Again, neonatally infected rats had memory impairments when administered LPS in adulthood, which were prevented by administering minocycline before adult LPS challenge (which also prevented increases in IL-1β mRNA). Microglia isolated from the fetuses of maternal sheep injected with LPS and then maintained in vitro for 3 weeks, had significantly higher production of IL-1β (>4-fold) in response to LPS in vitro relative to controls.45 The expression of heme oxygenase (decycling) 1, which is thought to have an anti-inflammatory role,46 was strongly downregulated in LPS/LPS microglia.
Training of innate immune cells by non-microbial stimuli
Upon cellular damage or stress, host cells release a variety of molecules. Some of these released molecules can activate PRR and initiate an inflammatory/immune response, much like PAMP. These endogenous PRR-stimulating non-microbial molecules are known as danger-associated molecular patterns (DAMPs).47 Examples of DAMPs include high-mobility group box-1, S-100 proteins, heat shock proteins, hyaluronan, surfactant protein, IFN-α, uric acid, fibronectin, beta defensin and cardiolipin, amongst others.47 Many of these DAMPs are oxidized versions of proteins and other molecules present on apoptotic host cells or in cellular debris.48 These molecules are oxidized as a result of cellular damage/death-induced reactive oxygen species generation, resulting in hydrophobic regions being exposed and available to be recognized by PRR as DAMP.47 Many of these endogenous oxidized molecules and hydrophobic regions share molecular identity with microbial PAMP.47, 48 Thus, we might expect DAMP to be also capable of inducing training. Exposure of human monocytes to oxidized low-density lipoprotein (1–10 μg ml−1) for 24 h, but not low-density lipoprotein, for example, results in increased protein production (up to fivefold) of TNF-α, IL-6 and IL-8, upon re-stimulation 7 days later with various TLR-4 (for example, LPS) and TLR-2 agonists (for example, Pam3Cys).49 Blocking these TLRs or their downstream pathways inhibited the training effects.
The study above is the only study that we are aware that has directly investigated whether DAMP can induce training. There are, however, a few studies that provide indirect evidence for DAMP-induced training. Sterile traumatic brain injury (TBI) releases a suite of DAMP,50 and might therefore be a systemic non-infectious immunostimulatory event capable of inducing microglial training. In humans, TBI is associated with microglial activation on positron emission tomography (PET) decades later,51 and is a risk factor for depression.52 Microglia isolated from mice that had been injected peripherally with LPS 1 month after TBI showed increased IL-1β and TNF-α mRNA expression relative to microglia from LPS exposed only mice.53 This heightened microglial activity was accompanied by depressive-like behaviors in the mice. Although suggestive, this is not direct evidence for DAMP-induced microglial training however, as TBI is a complex multifaceted phenomenon and the microglia were not stimulated ex vivo, thus raising the possibility that interactions with other cell types may have been responsible for the upregulation of microglial proinflammatory function. Microglia isolated from mice 24 days after repeated social defeat stress also had higher IL-1β, IL-6 and TNF-α mRNA expression, and IL-1-β protein production in response to ex vivo LPS stimulation relative to non-stressed controls.27 This upregulation in microglial proinflammatory function was accompanied by an increase in social avoidance behavior in repeated social defeat stress mice. Whether, specifically, stress-induced release of DAMPs resulted in microglial training was not investigated however. Psychological and physical stress results in the release of a variety of DAMP both peripherally and centrally, including heat shock protein 72, uric acid and high-mobility group box-1.54, 55, 56 Stressor exposure also releases microbial-associated molecular patterns (MAMPs) from the gut microbiota into the blood and/or extracellular environment.57 It seems likely that DAMP and MAMP, like PAMP, are capable of training innate immune cells. Given the strong association between stress, inflammation and neuropsychiatric illnesses, investigating whether stress-induced release of DAMP or MAMP train microglia in a homogeneous and heterogeneous manner is an intriguing avenue of future study.
Mechanisms underlying training
Several studies have specifically investigated whether training is underwritten by epigenetic remodeling. Human monocyte training as a result of BCG or Candida/β-glucan exposure results in an increase in H3K4me3, a histone modification associated with the regulation of immune-related genes, at the promoters of target genes, including TNF-α, IL-6 and IL-18.38 Consequently, blocking histone methylation by inhibition of histone methyltransferases prevented training. Training of monocytes by non-microbial stimuli, such as oxidized low-density lipoprotein, is also dependent on the enrichment of H3K4me3 at the promoters of various immune-related genes.49 β-glucan training in human monocytes has also been associated with epigenetic remodeling of various metabolic pathways.28, 58 β-glucan-trained monocytes display reduced oxygen consumption and increased glucose consumption, consistent with a switch from oxidative metabolism to glycolysis. This metabolic shift is characteristic of activated monocytes and macrophages, and, as a result, disrupting these changes in cellular metabolism inhibits training. Further, in mice, LPS-induced macrophage training results in phosphorylation of the stress-response transcription factor ATF7.59 ATF7 normally suppresses a group of genes encoding factors involved in innate immunity in macrophages by recruiting the histone H3K9 dimethyltransferase G9a. Training leads to the release of ATF7 from chromatin and a decrease in repressive histone H3K9me2 marks, and thus increased expression of immune-related genes. Interestingly, ATF7 is also phosphorylated as a result of social isolation stress and various other stressors.60
Whilst to our knowledge there have been no studies investigating whether epigenetic remodeling is responsible for training effects in microglia, there is evidence that epigenetic remodeling does underlie tolerance in microglia. In response to a high-dose in vivo LPS exposure, isolated mouse microglia displayed reduced proinflammatory cytokine production upon a secondary LPS stimulation in vitro.61 These effects were mediated by a reduction in the levels of H3K4me3 at the promoters of various immune-related genes. Whilst proinflammatory cytokine production was diminished, phagocytic activity and nitric oxide production were enhanced however. This study is interesting, not only because it demonstrates that microglia are capable of functional epigenetic remodeling but also because it shows that the dichotomy between innate immune tolerance versus training is potentially simplistic. Whilst some proinflammatory and neurotoxic innate immune cell functions may be downregulated, others may be simultaneously upregulated. As proinflammatory cytokines, reactive oxygen species and reactive nitrogen species all have the potential to be neurotoxic, it will be important to delineate the extent to which production of these different molecules are affected as a result of training.
Implications for neuropsychiatric diseases
There remain a lot of unanswered questions regarding the details of trained innate immunity. We have listed some of these in Table 2. Most important, given the association between neuroinflammation and psychiatric disease, is whether trained innate immunity contributes to the onset and progression of neuropsychiatric disorders. A key unknown is also the duration for which training can last. That training in human monocytes has been detected up to a year post primary exposure suggests that training effects can be long-term. This is an important observation. Early-life immunostimulatory events such as stress and infection are risk factors for the development of neuropsychiatric disorders, and possibly neurodegenerative disorders, in adulthood.62 Indeed, children prenatally exposed to elevated levels of the stress hormone cortisol reported persistent changes in the innate immunity with higher level of proinflammatory cytokines and subsequent development of emotional, behavioral and cognitive psychopathologies later in life.63, 64, 65, 66 Similarly, mice prenatally exposed to a bacterial infection with LPS showed depression- and anxiety-related behaviors during adulthood. In addition, LPS-treated mice had reduced serotonin and noradrenaline levels in the hippocampus, a brain region very well known for its neurogenenic properties associated with cognitive and memory functioning.67 Therefore, evidence seems to suggest that specific long-term changes in the inflammatory environment is alone sufficient for the occurrence of several psychopathologies later in life. One way in which these events are thought to contribute to the development of neuropsychiatric illnesses is through the induction or a proinflammatory predisposed phenotype, which can lead to neuroinflammation, in particular following a subsequent, temporally distant, immunostimulatory event. This ultimately results in the disruption of key processes involved in central nervous system development and regulation throughout adolescence and adulthood.62 These secondary immunostimulatory events can be completely unrelated in nature to the primary immunostimulatory event. In animal models, for example, the combination of prenatal polyI:C (which activates TLR-3 (ref. 68)) followed by peripubertal stress,69 or TBI followed by LPS,53 induce depressive-like behaviors. These findings cannot be easily explained by adaptive immunity or classical understandings of innate immunity. That trained innate immunity is heterogeneous in nature, likely because different PAMP and DAMP can activate the same PRR or downstream signaling pathways, resulting in the reprogramming of similar epigenetic pathways, offers a potential mechanistic explanation. Adult microglia arise almost exclusively from a founding population of bone marrow-derived primitive macrophages during development and are maintained throughout life.70 Events early in life may lead to microglia later in life having a lower threshold for activation and/or the enhanced production of proinflammatory molecules that can cause impairments in neuroplasticity and neurogenesis.
Furthermore, after accounting for genetic factors, it is not clear why some individuals develop inflammation and psychiatric illness upon exposure to the same environmental risk factors whilst others do not.71 Trained innate immunity, specifically individual variation in the epigenetic landscape for immune-related genes and pathways as a result of previous inflammatory events, might account for some of the variation in how individuals respond to risk factors. It is possible that the presence of one inflammatory risk factor may train the innate immune system to react more vigorously in response to a subsequent, temporally distant, or even contemporaneous, inflammatory risk factor.
Once cells of the innate immune system such as microglia are trained, this could lead to a cycle in which (neuro)inflammation begets (neuro)inflammation (Figure 1). (Neuro)inflammation can result in cell damage and death, and thus the release of numerous DAMP. As suggested, DAMPs may themselves be capable of inducing training. Thus, as a result of infection or stress-induced (neuro)inflammation and cell injury/death, for example, the release of DAMP may train the innate immune system toward a proinflammatory phenotype, even if the initial (neuro)inflammatory insult did not. Upon secondary challenge at a distant time, the enhanced release of proinflammatory and neurotoxic molecules can then result in greater (neuro)inflammation, cell death and DAMP release, and so on (Figure 1). This may explain, for example, how prior episodes of depression, for example, appear to sensitize immune responses to subsequent depressive episodes, with levels of proinflammatory cytokines and chemokines increasing with sequential depressive episodes.72, 73 Indeed, several studies have found that baseline depression and/or anxiety, even subclinical, predict a later development of depression in hepatitis C virus patients receiving the standard treatment with the proinflammatory cytokine IFN-α.74, 75, 76 One plausible hypothesis for such phenomenon is that upon primary challenge with the virus, DAMP recognition by PRRs elicit antimicrobial responses that regulate facets of the innate immune system,28 which potentially predispose to the occurrence of a first episode of depression. In addition, such DAMP-mediated training immunity implicates regulation of monocytes to macrophages differentiation,28 which upon secondary challenge with IFN-α treatment may ultimately predispose the individual to a second episode of depression. Although several studies reported that a history of past psychiatric disturbance significantly increased the risk of depression during IFN-α therapy,18 on the other hand other studies did not find that a past history of depression significantly increases the risk of neuropsychiatric disturbance.74, 77, 78, 79 This suggests that having a blunted response is also plausible and that the release of DAMP in presence of (neuro)inflammation could theoretically result in tolerance. However, whether tolerance or training occurs may well be dependent on the nature, doses, duration and frequency of immunostimulatory events.

Trained innate immunity and a resulting neuroinflammatory cycle. Red represents an immune stimulation and blue represents a consequence. The release of danger-associated molecular pattern (DAMP) is a consequence of neuronal injury and death, but can serve as a potential secondary immune stimulation, as well as a reinforcing stimulation for pre-existing training.
Conclusion
Many questions relating to the contribution of trained innate immunity to disease onset and progression in neuropsychiatric disorders need to be addressed. The discovery of trained innate immunity has opened up a new dimension in the field of immunotherapeutics, such as vaccine development. It is likely that trained innate immunity will also have a significant impact on our understanding of disorders in which the innate immune system is implicated, including neuropsychiatric illnesses, as well as the development of novel treatments for such disorders.
References
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB . Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2015; 2: 258–270.
Jeon SW, Kim YK . Neuroinflammation and cytokine abnormality in major depression: Cause or consequence in that illness? World J Psychiatry 2016; 6: 283–293.
Aricioglu F, Ozkartal CS, Unal G, Dursun S, Cetin M, Müller N . Neuroinflammation in schizophrenia: a critical review and the future. Klinik Psikofarmakoloji Bulteni 2016; 26: 429–437.
Jones KA, Thomsen C . The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci 2013; 53: 52–62.
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O . Neuroinflammation and psychiatric illness. J Neuroinflammation 2013; 10: 43.
Lull ME, Block ML . Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010; 7: 354–365.
Borsini A, Zunszain PA, Thuret S, Pariante CM . The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci 2015; 38: 145–157.
Bakunina N, Pariante CM, Zunszain PA . Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology 2015; 144: 365–373.
Streit WJ, Mrak RE, Griffin WST . Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 2004; 1: 14.
Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC et al. The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol 2012; 92: 959–975.
Perry VH, Nicoll JAR, Holmes C . Microglia in neurodegenerative disease. Nat Rev Neurol 2010; 6: 193–201.
Yirmiya R, Rimmerman N, Reshef R . Depression as a microglial disease. Trends Neurosci 2015; 38: 637–658.
Soczynska JK, Mansur RB, Brietzke E, Swardfager W, Kennedy SH, Woldeyohannes HO et al. Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav Brain Res 2012; 235: 302–317.
Zheng X, Zhang X, Wang G, Hao H . Treat the brain and treat the periphery: toward a holistic approach to major depressive disorder. Drug Discov Today 2015; 20: 562–568.
Delves PJ, Martin SJ, Burton DR, Roitt IM . Roitt's Essential Immunology. John Wiley & Sons, 2011.
Netea MG, Latz E, Mills KHG, O'Neill LAJ . Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol 2015; 16: 675–679.
Netea MG, Quintin J, van der Meer JWM . Trained Immunity: a memory for innate host defense. Cell Host Microbe 2011; 9: 355–361.
van 't Wout JW, Poell R, van Furth R . The role of BCG/PPD-activated macrophages in resistance against systemic candidiasis in mice. Scand J Immunol 1992; 36: 713–719.
Bistoni F, Verducci G, Perito S, Vecchiarelli A, Puccetti P, Marconi P et al. Immunomodulation by a low-virulence, agerminative variant of Candida albicans. Further evidence for macrophage activation as one of the effector mechanisms of nonspecific anti-infectious protection. J Med Vet Mycol 1988; 26: 285–299.
Condliffe AM, Kitchen E, Chilvers ER . Neutrophil priming: pathophysiological consequences and underlying mechanisms. Clin Sci 1998; 94: 461–471.
Netea MG . Training innate immunity: the changing concept of immunological memory in innate host defence. Eur J Clin Invest 2013; 43: 881–884.
Schroder K, Sweet MJ, Hume DA . Signal integration between IFNγ and TLR signalling pathways in macrophages. Immunobiology 2006; 211: 511–524.
Perry VH, Holmes C . Microglial priming in neurodegenerative disease. Nat Rev Neurol 2014; 10: 217–224.
Ransohoff RM, Perry VH . Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol 2009; 27: 119–145.
Wynne AM, Henry CJ, Godbout JP . Immune and behavioral consequences of microglial reactivity in the aged brain. Integr Comp Biol 2009; 49: 254–266.
Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LAB, Jacobs C et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J Innate Immun 2014; 6: 152–158.
Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF . Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immun 2015; 46(C): 212–220.
Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014; 345: 1251086–1251086.
Poli A, Kmiecik J, Domingues O, Hentges F, Blery M, Chekenya M et al. NK cells in central nervous system disorders. J Immunol 2013; 190: 5355–5362.
Akira S, Uematsu S, Takeuchi O . Pathogen recognition and innate immunity. Cell 2006; 124: 783–801.
Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest 2000; 105: 497–504.
Biswas SK, Lopez-Collazo E . Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol 2009; 30: 475–487.
Martin SF . Adaptation in the innate immune system and heterologous innate immunity. Cell Mol Life Sci 2014; 71: 4115–4130.
Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol 2014; 21: 534–545.
Tribouley J, Tribouley-Duret J, Appriou M . Effect of Bacillus Callmette Guerin (BCG) on the receptivity of nude mice to Schistosoma mansoni. C R Seances Soc Biol Fil 1978; 172: 902–904.
Bistoni F, Vecchiarelli A, Cenci E, Puccetti P, Marconi P, Cassone A . Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infect Immun 1986; 51: 668–674.
Jensen KJ, Larsen N, Biering-Sørensen S, Andersen A, Eriksen HB, Monteiro I et al. Heterologous immunological effects of early BCG vaccination in low-birth-weight infants in Guinea-Bissau: a randomized-controlled trial. J Infect Dis 2015; 211: 956–967.
Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012; 12: 223–232.
Püntener U, Booth SG, Perry VH, Teeling JL . Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation 2012; 9: 146.
Bilbo SD . Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci 2005; 25: 8000–8009.
Bland ST, Beckley JT, Watkins LR, Maier SF, Bilbo SD . Neonatal Escherichia coli infection alters glial, cytokine, and neuronal gene expression in response to acute amphetamine in adolescent rats. Neurosci Lett 2010; 474: 52–57.
Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile Virus infection. PLoS Pathog 2012; 8: e1003039–16.
Chauhan VS, Sterka DG Jr., Furr SR, Young AB, Marriott I . NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia 2009; 57: 414–423.
Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD . Microglia and memory: modulation by early-life infection. J Neurosci 2011; 31: 15511–15521.
Cao M, Cortes M, Moore CS, Leong SY, Durosier LD, Burns P et al. Fetal microglial phenotype in vitro carries memory of prior in vivo exposure to inflammation. Front Cell Neurosci 2015; 9: 12.
Ye M, Wang Q, Zhang W, Li Z, Wang Y, Hu R . Oroxylin A exerts anti-inflammatory activity on lipopolysaccharide-induced mouse macrophage via Nrf2/ARE activation. Biochem Cell Biol 2014; 92: 337–348.
Seong SY, Matzinger P . Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 2004; 4: 469–478.
Matt U, Sharif O, Martins R, Knapp S . Accumulating evidence for a role of oxidized phospholipids in infectious diseases. Cell Mol Life Sci 2014; 72: 1059–1071.
Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP . Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol 2014; 34: 1731–1738.
Manson J, Thiemermann C, Brohi K . Trauma alarmins as activators of damage-induced inflammation. Br J Surg 2012; 99 (Suppl 1): 12–20.
Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM et al. Inflammation after trauma: Microglial activation and traumatic brain injury. Ann Neurol 2011; 70: 374–383.
Jorge RE, Robinson RG, Moser D, Tateno A, Crespo-Facorro B, Arndt S . Major depression following traumatic brain injury. Arch Gen Psychiatry 2004; 61: 42–50.
Fenn AM, Gensel JC, Huang Y, Popovich PG . Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 2014; 76: 575–584.
Fleshner M, Campisi J, Amiri L, Diamond DM . Cat exposure induces both intra- and extracellular Hsp72: the role of adrenal hormones. Psychoneuroendocrinology 2004; 29: 1142–1152.
Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF . Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 2015; 35: 316–324.
Maslanik T, Mahaffey L, Tannura K, Beninson L, Greenwood BN, Fleshner M . The inflammasome and danger associated molecular patterns (DAMPs) are implicated in cytokine and chemokine responses following stressor exposure. Brain Behav Immun 2013; 28(C): 54–62.
Fleshner M . Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun 2013; 27(C): 1–7.
Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V et al. mTOR- and HIF-1 -mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014; 345: 1250684–1250684.
Yoshida K, Maekawa T, Zhu Y, Renard-Guillet C, Chatton B, Inoue K et al. The transcription factor ATF7 mediates lipopolysaccharide-induced epigenetic changes in macrophages involved in innate immunological memory. Nat Immunol 2015; 16: 1034–1043.
Maekawa T, Kim S, Nakai D, Makino C, Takagi T, Ogura H et al. Social isolation stress induces ATF-7 phosphorylation and impairs silencing of the 5-HT 5B receptor gene. EMBO J 2009; 29: 196–208.
Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun 2015; 48(C): 205–221.
Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 2014; 10: 643–660.
Talge NM, Neal C, Glover V, Early Stress TR Prevention Science Network F, Neonatal Experience on Cet alAntenatal maternal stress and long-term effects on child neurodevelopment: how and why? J Child Psychol Psychiatry 2007; 48: 245–261.
Glover V . Annual research review: prenatal stress and the origins of psychopathology: an evolutionary perspective. J Child Psychol Psychiatry 2011; 52: 356–367.
Van den Bergh BRH, Van Calster B, Smits T, Van Huffel S, Lagae L . Antenatal maternal anxiety is related to HPA-axis dysregulation and self-reported depressive symptoms in adolescence: a prospective study on the fetal origins of depressed mood. Neuropsychopharmacology 2007; 33: 536–545.
Sandman CA, Buss C, Head K, Davis EP . Fetal exposure to maternal depressive symptoms is associated with cortical thickness in late childhood. Biol Psychiatry 2015; 77: 324–334.
Depino AM . Early prenatal exposure to LPS results in anxiety- and depression-related behaviors in adulthood. Neuroscience 2015; 299: 56–65.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA . Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001; 413: 732–738.
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013; 339: 1095–1099.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–845.
Berk M, Williams LJ, Jacka FN, Neil AO, Pasco JA, Moylan S et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med 2013; 11: 1–1.
Maes M, Mihaylova I, Kubera M, Ringel K . Activation of cell-mediated immunity in depression: association with inflammation, melancholia, clinical staging and the fatigue and somatic symptom cluster of depression. Prog Neuropsychopharmacol Biol Psychiatry 2012; 36: 169–175.
Celik C, Erdem M, Caycı T, Ozdemir B, Akgul EO, Kurt YG et al. The association between serum levels of neopterin and number of depressive episodes of major depression. Prog Neuropsychopharmacol Biol Psychiatry 2010; 34: 372–375.
Hauser P, Khosla J, Aurora H, Laurin J, Kling MA, Hill J et al. A prospective study of the incidence and open-label treatment of interferon-induced major depressive disorder in patients with hepatitis C. Mol Psychiatry 2002; 7: 942–947.
Dieperink E, Ho SB, Thuras P, Willenbring ML . A prospective study of neuropsychiatric symptoms associated with interferon-a-2b and ribavirin therapy for patients with chronic hepatitis C. Psychosomatics 2011; 44: 104–112.
Capuron L, Ravaud A . Prediction of the depressive effects of interferon alfa therapy by the patient's initial affective state. N Engl J Med 1999; 340: 1370–1370.
Renault PF, Hoofnagle JH, Park Y . Psychiatric complications of long-term interferon alfa therapy. Arch Intern Med 1987; 147: 1577–1580.
Schaefer M . Adherence and mental side effects during hepatitis C treatment with interferon alfa and ribavirin in psychiatric risk groups. Hepatology 2003; 37: 443–451.
Pariante CM, Orrù MG, Baita A, Farci MG, Carpiniello B . Treatment with interferon-α in patients with chronic hepatitis and mood or anxiety disorders. Lancet 1999; 354: 131–132.
Acknowledgements
APS is supported by a National Institute for Health Research (NIHR) Biomedical Research Centre-Francis Crick Institute Clinical Training Fellowship. PAZ and AB are supported by the NIHR Maudsley Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PAZ has received research funding from Johnson & Johnson as part of a program of research on depression and inflammation, and from the Medical Research Council (UK) and the Wellcome Trust for research on depression and inflammation as part of two large consortia that also include Johnson & Johnson, GSK, and Lundbeck. The remaining authors declare no conflict of interest.
PowerPoint slides
Rights and permissions
About this article

Cite this article
Salam, A., Borsini, A. & Zunszain, P. Trained innate immunity: a salient factor in the pathogenesis of neuroimmune psychiatric disorders. Mol Psychiatry 23, 170–176 (2018). https://doi.org/10.1038/mp.2017.186
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2017.186
This article is cited by
-
Immunophenotypes in psychosis: is it a premature inflamm-aging disorder?
Molecular Psychiatry (2024)
-
Cortical microinfarcts potentiate recurrent ischemic injury through NLRP3-dependent trained immunity
Cell Death & Disease (2024)
-
Advancing preclinical models of psychiatric disorders with human brain organoid cultures
Molecular Psychiatry (2023)
-
Immune memory in the brain
Nature (2018)
