
Abstract
Neurodevelopmental disorders are multi-faceted and can lead to intellectual disability, autism spectrum disorder and language impairment. Mutations in the Forkhead box FOXP1 gene have been linked to all these disorders, suggesting that it may play a central role in various cognitive and social processes. To understand the role of Foxp1 in the context of neurodevelopment leading to alterations in cognition and behaviour, we generated mice with a brain-specific Foxp1 deletion (Nestin-CreFoxp1−/−mice). The mutant mice were viable and allowed for the first time the analysis of pre- and postnatal neurodevelopmental phenotypes, which included a pronounced disruption of the developing striatum and more subtle alterations in the hippocampus. More detailed analysis in the CA1 region revealed abnormal neuronal morphogenesis that was associated with reduced excitability and an imbalance of excitatory to inhibitory input in CA1 hippocampal neurons in Nestin-CreFoxp1−/− mice. Foxp1 ablation was also associated with various cognitive and social deficits, providing new insights into its behavioural importance.
Similar content being viewed by others

Introduction
Dysfunction of motor, social, sensory and cognitive aspects play a major role in autism spectrum disorder (ASD) and intellectual disability (ID). A high comorbidity is often observed between these disorders, suggesting that mutations in critical genes can cause a spectrum of neuropsychiatric phenotypes.1 The Forkhead box transcription factor FOXP1, for example, has been linked to various cognitive disorders. FOXP1-specific deletions, mutations and chromosomal breakpoints interrupting the gene have been reported in patients with ID, ASD, speech and language deficits, and motor development delay.1, 2, 3, 4, 5 Collectively, these studies have provided strong evidence for FOXP1 mutations underlying specific cognitive phenotypes; however, the importance of FOXP1 in brain function remains largely undefined.
FOXP1 is a member of the Forkhead Box P (FOXP) subfamily of transcription factors, which also includes FOXP2.6 The role of FOXP2 in human speech and language has generated extreme interest 7 and the emerging evidence now implicating FOXP1 in the pathology of language impairment as well as a broader range of human cognitive disorders is intriguing. FOXP1 and FOXP2 form heterodimers for transcriptional regulation8 and are co-expressed in many brain regions,9,10 suggesting that they co-operate in common pathways for cognitive and language development.
Animal models often pave the way to understanding a human disorder at the causal and mechanistic level. Several mutant and knockout (KO) Foxp2 mouse models have been generated to elucidate the role of Foxp2 in brain development, revealing insightful phenotypes including developmental delay, motor impairment, cerebellar abnormalities and disrupted synaptic plasticity in the striatum.11, 12, 13 These studies have helped to establish a role for Foxp2 in the development of neural circuits contributing to language development and possibly wider cognitive function. Animal models are not yet available to investigate the role of Foxp1 in brain development, although a conventional Foxp1 KO mouse, which is lethal at embryonic day (E) 14 because of a cardiac defect,14 has illustrated the importance of Foxp1 in a range of non-neural developmental processes15 and in the development of motor neurons in the spinal cord.16
In this study, we report the generation and characterisation of mice where Foxp1 is deleted specifically in the brain. Nestin-CreFoxp1−/−mice are viable and hence are suitable to address Foxp1 function in brain development.
Materials and Methods
Generation of a conditional Foxp1 KO mouse
Homozygous floxed Foxp1 mice17 were crossed with Nestin-Cre deleter mice18 heterozygous for the floxed Foxp1 allele, producing 25% homozygous Foxp1 KO, 25% heterozygous Foxp1 KO and 50% wild-type (WT) offspring. All mice were C57 Bl6.
Immunohistochemistry and quantification of striatal region
Immunohistochemistry was performed on paraffin sections of E14, E16 and E18, postnatal day (P) 1 and 21, and adult brains (see Supplementary information). Striatal area was quantified using the freehand tool in ImageJ (US National Institutes of Health, Bethesda, MD, USA). Tyrosine hydroxylase staining was used to define the striatal area for quantification. The striatal area was normalised to the total brain area.
Affymetrix mRNA microarray
Tissue preparation (P1 striata), RNA isolation, gene expression profiling and data analysis was performed as described in the Supplementary information.
Electrophysiological analysis of CA1 hippocampal pyramidal neurons
Electrophysiological analyses were performed on 250 μm transverse hippocampal slices of Foxp1 KO and WT mice aged between P18 and P25 (see Supplementary information).
Behavioural analyses of Foxp1 KO mice
A total of 49 adult male mice aged between 8 and 18 weeks (WT: n=34; Foxp1 KO: n=15) were used in behavioural tests and animals were age-matched as much as possible for each individual test. Tests for locomotor activity, repetitive behaviours, non-spatial and spatial short-term memory, anxiety, social interaction, prepulse inhibition (PPI) of the acoustic startle reflex (ASR), and nestbuilding behaviour were performed and analysed as described in the Supplementary information. Social interaction was assessed in an open field apparatus with a same-sex juvenile partner.
Results
We deleted Foxp1 specifically in neural tissues using the Cre-Lox system to generate a conditional Foxp1 mutant mouse, Nestin-CreFoxp1−/−. Immunohistochemistry confirmed a loss of Foxp1 protein in the brains of Nestin-CreFoxp1−/−mice (Supplementary Figure 1). Nestin-CreFoxp1−/− mice are referred to as Foxp1 KO mice throughout the manuscript. Foxp1 KO mice are viable but have a significantly reduced body weight compared with WT littermates (see Supplementary information).
Analysis of Foxp1 KO brain morphology, neuronal morphogenesis and affected pathways in the striatum
The striatum is significantly disrupted in the brains of Foxp1 KO mice
To determine whether loss of Foxp1 causes gross morphological abnormalities, we first performed Calbindin immunohistochemistry (Figure 1a) and Nissl stainings (Figure 2) on adult Foxp1 KO brains. A significant enlargement of the lateral ventricles in Foxp1 KO brains was observed (WT=0.00974 μ m−2; KO=0.0504 μ m−2, P-value (t test)=<0.0001; values represent lateral ventricle area normalised to the area of the whole brain), comparable with the magnetic resonance imaging scan of a patient with haploinsufficiency of FOXP1, which revealed prominent lateral ventricles.5 In addition, a striking reduction of the striatal region (Figures 1a, b and c) was observed. To investigate the striatal phenotype further, we stained the striatum of adult brains using tyrosine hydroxylase immunohistochemistry, which revealed a reduced dorsal striatum and an enlargement of the ventral region (Figure 1b). The distinction between the dorsal and ventral striatum is based on specific cortical, thalamic and dopaminergic inputs and is not defined by a distinct border.19 As no markers exist that uniquely characterise either region, we quantified the total striatal area (defined as the area of tyrosine hydroxylase positive staining), which was significantly smaller in Foxp1 KO brains compared with WT (Figure 1c).

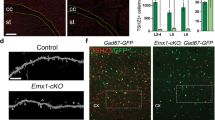
Morphological defects in the developing Foxp1 KO brain. Calbindin (a) and tyrosine hydroxylase (b) immunohistochemistry showing gross morphological disruption in the striatal region. Scale bar represents 1000 μm in adult brain sections, 500 μm in all others. (c) Quantification of the striatal region as defined by area of tyrosine hydroxylase positive staining, demonstrating a significant reduction in the striatum of Foxp1 KO brains, starting at P1. At least 12 sections from at least three WT and three KO brains were quantified for each stage. (d) Pathway analysis of microarray expression studies on P1 Foxp1 KO and WT striatal tissue showing the top 11 significantly regulated pathways.

Nissl staining on adult brain slices showing morphological alteration in the CA1 hippocampal region of KO brains compared with WT ( × 4) (a). (b) Higher magnification images of the CA1 region ( × 20). At least four brains were examined for each genotype. Scale bars represent 500 μm.
To determine when the striatal phenotype arises, we examined the striatum at earlier stages of development. At P21 (Figures 1a and b; third panels), the striatum was as affected as in adult brains, but at P1 (Figures 1a and b; second panels), striatal disruption was considerably more subtle (Figure 1c). At E18, no difference was detected (Figure 1a and b; upper panels, 1c). These findings suggest that Foxp1 plays an important role in normal development of the striatum starting at early postnatal stages.
Foxp1 regulates pathways associated with cell proliferation in the early postnatal striatum
To define those pathways involving Foxp1 in the striatum at early postnatal stages, we performed microarray expression studies comparing gene expression in striatal tissue from Foxp1 KO and WT animals at P1. We identified 85 significantly regulated genes, of which 61 were upregulated and 24 were downregulated (Supplementary Table 1). Gene Ontology and pathway analysis of the putative target genes was performed and revealed that pathways involved in nucleosome and chromatin assembly, mitosis and DNA replication are significantly affected (11 significantly altered pathways of 556 pathways analysed; Figure 1d).
Foxp1 KO striatal neurons show increased dendritic branching in vitro
To examine the morphology of individual Foxp1 KO striatal neurons, we transfected primary striatal neurons cultured from E15 brains with a plasmid encoding eGFP (Supplementary Figure 2A). Sholl analysis revealed that Foxp1 KO striatal neurons have a significantly more elaborate dendritic arbor than WT neurons (Supplementary Figure 2B). The difference between the groups remained significant after correction for multiple testing using Bonferroni correction.
Morphological and electrophysiological analysis of the Foxp1 KO hippocampus
Subtle alterations in the CA1 region of the hippocampus in Foxp1 KO mice
Nissl stainings on adult brain revealed a misalignment of cells from the CA1 region of the hippocampus that appeared to be less densely packed compared with WT (Figure 2). CA1 hippocampal pyramidal neurons express Foxp1 but not the closely related Foxp2 (Supplementary Figure 3). These findings prompted us to perform whole-cell current-clamp recordings in neurons within the CA1 region of the hippocampus.
Foxp1 KO CA1 hippocampal pyramidal neurons have reduced excitability
Whole-cell patch-clamp recordings of individual pyramidal neurons within the CA1 region of the hippocampus at P21 were measured and revealed a significantly reduced firing rate in response to depolarizing current steps in Foxp1 KO neurons (Figures 3a and b). The membrane capacitance, input resistance and resting membrane potential were unaffected (Figure 3c). Recordings of spontaneous miniature excitatory postsynaptic currents revealed significantly increased amplitudes in Foxp1 KO mice, while the frequency remained unchanged (Figures 3d and e). Foxp1 is expressed postsynaptically in CA1 neurons but not presynaptically in CA3 neurons, suggesting an increased number of synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in Foxp1 KO neurons. The absence of presynaptic changes was further confirmed by measuring the coefficient of variation and paired-pulse ratio of postsynaptic currents evoked by Schaffer collateral stimulation. The coefficient of variation is a measure of the fluctuation of the postsynaptic response which is solely determined by the presynaptic release probability and the number of release sites. The paired-pulse ratio is a quantification of the postsynaptic response facilitation that is observed for the second of two stimulation pulses applied in close succession and is a function of the release probability. Both coefficient of variation and paired-pulse ratio remained unaltered in Foxp1 KO neurons (Supplementary Figure 4), confirming that the increase in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor-mediated currents is purely postsynaptic in CA1 neurons. The amplitude and frequency of miniature inhibitory postsynaptic currents was similar in control and Foxp1 KO mice (Figures 3f and g). Field recording experiments showed that tetanus-induced long-term potentiation at Shaffer collateral CA1 synapses was also similar in WT and Foxp1 KO mice (Supplementary Figure 5). Sholl analysis of CA1 pyramidal cells revealed that apical dendritic branches in Foxp1 KO neurons did not exceed 600 μm from the soma and clustered in closer proximity to the somatic compartment than in WT neurons. This difference was statistically significant (Supplementary Figure 6 A, B, C). Basal dendrite length and total apical dendrite length were not affected.

Decreased excitability and increased excitatory synaptic transmission in Foxp1 KO hippocampal pyramidal neurons. (a) Sample spike trains evoked by a 300 pA somatic current injection in a control (left) and a Foxp1 KO CA1 pyramidal neuron (right). (b) mean f-i curve for control (n=10) and Foxp1 KO neurons (n=12). ***P<0.001 (ANOVA, main effect). (c) Cell capacitance (Cm), cell resistance (Rm) and resting membrane voltage (Vrest) in control and Foxp1 KO neurons. (d) Sample traces of miniature EPSCs from WT and Foxp1 KO cells recorded in voltage clamp at −70 mV. Although the miniature excitatory postsynaptic currents peak frequency was not significantly altered, the mean peak amplitude was significantly larger in Foxp1 KO neurons (e, two-tailed t-test. *P<0.05). WT, n=12/3 cells/mice; Foxp1 KO, n=10/2 cells/mice. (f) Miniature inhibitory postsynaptic current example traces from each genotype. There was no significant change in the mean miniature inhibitory postsynaptic current peak frequency and amplitude between control (n=13/2 cells/mice) and Foxp1 KO cells (n=11/2 cells/mice). (g) Data are shown as mean±s.e.m.
Behavioural analysis of Foxp1 KO mice
We were interested to examine the behavioural phenotype of our Foxp1 KO mice as FOXP1 mutations are associated with various behavioural deficits in humans, including social unattainability, hyperactivity, altered learning and memory, and specific obsessions.2,3 We analysed Foxp1 heterozygous mice in all tests described, except for repetitive behaviour and anxiety. Foxp1 heterozygous KO mice performed equally to WT mice in all tests (data not shown).
Hyperactivity in Foxp1 KO mice
We first assessed the locomotor ability of the mice. For both distance travelled and total rearing time, Foxp1 KO mice displayed a higher activity level within the open field than WT mice (Figures 4a and b) (distance travelled P<0.001; total rearing time P=0.011) of the mice in the open field, suggesting that Foxp1 KO mice are hyperactive.

Altered behaviours in Foxp1 KO mice. (a, b) Locomotor activity as measured by distance travelled (a) and total rearing time (b). (c) Spatial short-term memory measured by object location test. (d) Analysis of non-spatial short-term memory by object discrimination test. (e) Analysis of repetitive behaviours illustrating the total frequency of jackhammer jumping, jumping and upright wall scrabbling. (f–g) Tests of social interaction in Foxp1 KO mice including social exploration (f) and social retreat (g). (h) Measurement of prepulse inhibition (PPI) of the acoustic startle reflex (ASR). In all graphs, black bars represent WT and orange bars represent KO values. Fourteen WT and 6 Foxp1 KO mice were analysed in each test, except repetitive behaviour, where 11 WT and 6 Foxp1 KO animals were analysed.
Impaired short-term memory in Foxp1 KO mice
Spatial and non-spatial short-term memory was measured in the open field using the object location and object recognition test. Exploration of the objects was slightly enhanced in both tests in Foxp1 KO mice compared with WT mice during the initial exploration phase (P=0.054). However, Foxp1 KO mice failed to discriminate the novel object during object recognition testing (P=0.002) (Figure 4d) and the object placed in a novel position during object location testing (P=0.017) (Figure 4c), suggesting that Foxp1 KO mice have impaired spatial and non-spatial short-term memory.
Increased repetitive behaviours in Foxp1 KO mice
A first qualitative behavioural screen in the open field revealed that repetitive behaviours were almost absent in WT mice. In contrast, jumping and wall scrabbling was frequently observed in KO animals. Frequency (P<0.001), duration (P<0.001) and score for repetitive behaviours (P<0.001) were significantly increased in KO mice compared with WT (Figure 4e).
Impaired social behaviour in Foxp1 KO mice
Foxp1 KO mice showed strikingly reduced exploratory behaviour in all categories (anogenital exploration, P<0.001; non-anogenital exploration, P<0.001; approach/following, P<0.001) (Figure 4f, Supplementary videos 1 and 2). Additionally, Foxp1 KO mice showed a significantly increased evasion of social contact compared with WT (P<0.001) (Figure 4g). This reduced social exploratory behaviour is not attributable to a heightened anxiety as Foxp1 KO mice actually show reduced anxiety (see below). Although not classically a social behaviour, we also assessed the nestbuilding ability of the mice, which is considered important within a colony of animals. Nestbuilding ability of Foxp1 KO mice was drastically impaired, with no attempt made to construct a nest after nesting material was provided (P=0.008); (Supplementary figure 7C).
Foxp1 KO mice have altered PPI of the ASR
Startle is a fast response to sudden, intense stimuli. The ASR has been used as a behavioural tool to assess the neuronal basis of behavioural plasticity and investigate dysfunctions of sensorimotor information processing. PPI is the reduction of the ASR by presentation of an acoustic stimulus before the startling stimulus and is reduced in a variety of neuropsychiatric disorders characterised by a disruption in sensorimotor processing.20 As the striatum is involved in sensorimotor processing, we measured PPI of the ASR in Foxp1 KO mice.
The ASR amplitude of Foxp1 KO mice without a prepulse was lower than that of WT mice (WT ASR=79.45, s.e.m.=17.86; Foxp1 KO ASR=26.75, s.e.m.=3.96), possibly because of the reduced body weight of Foxp1 KO mice. However, this difference in ASR amplitude was not significant (P=0.074). Foxp1 KO mice showed a significantly reduced PPI than WT mice when exposed to a startle stimulus following a prepulse intensity of 70 dB and above (65 dB, P=0.338; 70 dB, P=0.002; 75 dB, P<0.001; 80 dB, P<0.001) (Figure 4h), suggesting impaired attentional processing. This difference is not attributable to the insignificant reduction in ASR amplitude as the PPI was calculated as a percent decrease of the ASR magnitude when the startle stimulus was preceded by a prepulse.
Reduced anxiety in Foxp1 KO mice
We observed a reduced frequency of anxiety-related behaviours in Foxp1 KO mice in the open field (Supplementary Figure 7A) and therefore decided to perform preliminary testing under low-anxiety conditions in the elevated plus maze. This revealed reduced anxiety in Foxp1 KO mice (values as means±s.e.m.: % time in open arms: KO=82.6±1.9; WT=43.8±15.0; P=0.11; % open arm entries: KO=80.6±5.2; WT=37.9±11.3; P=0.03). Therefore, a different cohort of animals was tested under high-anxiety-inducing conditions and this confirmed reduced anxiety in Foxp1 KO (Figure 4h). KO mice displayed significantly higher entries onto open arms (P=0.002) and time spent on open arms (P=0.006). No significant differences could be observed in closed arm entries (P>0.05) between both genotypes.
Discussion
Mutations in FOXP1 can lead to a spectrum of neurodevelopmental phenotypes including ID and ASD. Interestingly, the most striking frameshift and nonsense mutations were detected in those patients who presented an ASD phenotype.2,4 Here we used a Foxp1 KO mouse model to define the underlying neurodevelopmental pathology of the FOXP1 disorder and identified phenotypes that mirror those reported in both human patients and mouse models of cognitive disorders, particularly ASD.
Foxp1 KO behavioural phenotypes associated with ASD
Foxp1 KO mice have a reduced ability for short-term recognition memory and memory for spatial contexts, which have been described before in ASD patients21 and in mouse models of ASD.22, 23, 24 The effect on spatial memory may be explained by the CA1 hippocampal deficits we observed in Foxp1 KO as the hippocampus is important for spatial memory.25 The disruption of the striatal region in Foxp1 KO mice may also contribute to the deficits in learning and memory. It has been shown that striatal lesions and infusion of the striatum with a dopaminergic antagonist results in impaired performance in spatial learning tests,26 while object recognition is impaired by administration of glutamate antagonists to the striatum.27 Interestingly, the striatum has previously been associated with the pathology of ASD in both mice and humans.28,29
Foxp1 KO mice also displayed a higher occurrence of repetitive behaviours, in accordance with previous findings in mouse models of autism.22,28 Repetitive motor behaviour is associated with abnormal activation of dopaminergic cortical-basal ganglia circuitry30 and therefore might partially be explained by the morphological disruption we observed in the striatal region.
A striking reduction of social interest was also detected in Foxp1 KO mice. Difficulties communicating and interacting with other people is a key feature of human ASD, and reduced social interaction as well as hyperactivity has been reported in mouse models of ASD before.23,24,31 A strong PPI deficit was observed in Foxp1 KO mice, indicating impaired abilities for sensorimotor integration. Reduced PPI has been previously reported in ASD patients.32 This effect on PPI in Foxp1 KO mice may be partly explained by the reduction in the striatal region as a cortico-limbic-striatopallidal circuit is involved in the circuit regulating PPI.20
Imbalance of excitatory to inhibitory input in Foxp1 KO hippocampal neurons
Excitatory/inhibitory imbalance is a hallmark feature of ASD. Several studies have reported that ASD-related mutations selectively impact glutamatergic or GABAergic synapses without affecting the other, leading to an imbalance of excitatory and inhibitory inputs.33 We have shown that the amplitude of miniature excitatory postsynaptic currents but not miniature inhibitory postsynaptic currents is larger in Foxp1 KO CA1 hippocampal neurons, suggesting that Foxp1 KO neurons receive a disproportionate magnitude of excitatory to inhibitory input. In addition, excitability of CA1 pyramidal cells was reduced in Foxp1 KO mice. Whether these physiological deficits occur independently of each other is not clear. It is possible that the excitability of Foxp1 KO neurons is reduced to compensate for the increased glutamatergic transmission we observed, thus maintaining neurons within their physiological range of firing rate. Such a homeostatic scaling of neuronal excitability was observed, for instance, in response to changes in synaptic inputs in cultured cortical neurons.34
The role of Foxp1 in striatal development and neuronal morphogenesis
The striatal region is significantly reduced in early postnatal Foxp1 KO mice suggesting a role for Foxp1 in regulating striatal development. Comparison of gene expression in the Foxp1 KO and WT striatum suggested that Foxp1 may regulate the transcription of genes that induce proliferation of striatal neurons. Disruption of these pathways may explain the reduction in the Foxp1 KO striatum. Indeed, Foxp1 has previously been implicated in various types of cancer and depending on the tissue can function as both an oncogene and a tumour suppressor.6,35 Pathways involved in both the positive and negative regulation of programmed cell death were not found to be significantly affected in our expression analyses of P1 striatal tissue; therefore, impaired proliferation of striatal neurons may be the more likely explanation for the striatal phenotype.
Foxp1 may also play a role in neuronal morphogenesis. Our analyses revealed altered morphology of Foxp1 KO striatal neurons in vitro and CA1 pyramidal neurons in vivo. The closely related Foxp2 was shown already to regulate genes implicated in neurite outgrowth,36 suggesting that Foxp1 may also be involved in pathways that regulate morphogenesis in the embryonic brain, potentially as a heterodimer with Foxp2. Increased complexity of dendritic arborisation was also recently reported in striatal neurons in mice lacking Shank3 associated with ASD.28
Comparisons with the Foxp2 KO phenotype
FOXP1 and FOXP2 disruption causes distinct and overlapping clinical phenotypes, which have been reviewed previously.37 Although a core feature of the FOXP2 phenotype is developmental verbal dyspraxia, this has not been diagnosed in FOXP1 patients, who show more general language impairment together with additional cognitive phenotypes such as ID and ASD.37 FOXP1 and FOXP2 are biologically pleiotropic genes that may disrupt various intersecting developmental processes. It will be of extreme interest to define common and specific pathways regulated by these closely related transcription factors to further define the underlying pathology of the human phenotypes. Comparing the phenotypes of Foxp1 and Foxp2 mouse mutants represents important steps towards achieving this.
Homozygous loss of Foxp1 in all tissues is embryonically lethal because of a cardiac defect,14 whereas homozygous Foxp2 KO mice die around 2–3 postnatal weeks, possibly because of impaired lung function.11, 12, 13 We have now shown that brain-specific Foxp1 deletion results in viable homozygous offspring with neurodevelopmental defects, but it remains to be shown whether this is also true for Foxp2, as no conditional deletion mutant of Foxp2 exists to date.
Histological analysis of the early postnatal brain of homozygous Foxp2 KOs revealed an abnormal cerebellum, with reduced foliation.11, 12, 13 We observed no reduction of the cerebellum upon dissection of Foxp1 KO brains, which was expected as Foxp1 is not expressed in the cerebellum.9,10 Both Foxp1 and Foxp2, however, are strongly expressed in the developing and mature striatum,9,10 yet unlike Foxp1 homozygous KO mice, Foxp2 homozygotes showed no gross morphological abnormalities in the forebrain at P21.11, 12, 13 Although this implies that Foxp1 plays the more dominant role in early striatal development, compelling evidence exists that Foxp2 is important for normal development and function of the mature striatum. Neuroimaging analyses on members of the KE family with a point mutation in the Forkhead domain of Foxp2, which causes developmental verbal dyspraxia, have revealed alterations in the grey-matter density of the striatum38 and mice heterozygous for the identical point mutation in the KE family display impaired functioning of striatal circuits during the acquisition of motor skills.39 Therefore, Foxp1 may regulate striatal pathways as heterodimers together with Foxp2.
Foxp2 mutant mice do not produce ultrasonic isolation calls upon separation from their mother or nest.12,13 Such tests were beyond the scope of this study, but it will be of interest to determine whether Foxp1 KO mice show similar alterations in ultrasonic calls in the future. It is also conceivable that speech disruption in FOXP1 patients may be a consequence of the pronounced ID/ASD phenotype, rather than a disruption in specific language pathways.
In conclusion, our findings have demonstrated that Foxp1 is critical for multiple neurodevelopmental processes. Foxp1 appears to be crucial for normal cognitive function and social behaviour and plays an important role in striatal development at early postnatal stages. This is a time window when functional synapses are forming and the deficiencies associated with ASD may be initiated. The behavioural and physiological abnormalities in Foxp1 KO mice are consistent with ASD-like phenotypes in other mouse models of ASD, support findings in human patients with FOXP1 mutations and open up new opportunities for translational investigations.
References
Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic Boundaries. Cell 2012; 149: 525–537.
Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet 2010; 87: 671–678.
Horn D, Kapeller J, Rivera-Brugues N, Moog U, Lorenz-Depiereux B, Eck S et al. Identification of FOXP1 deletions in three unrelated patients with mental retardation and significant speech and language deficits. Hum Mutat 2010; 31: E1851–E1860.
O'Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011; 43: 585–589.
Le Fevre AK, Taylor S, Malek NH, Horn D, Carr CW, Abdul-Rahman OA et al. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A 2013; 161: 3166–3175.
Banham AH, Beasley N, Campo E, Fernandez PL, Fidler C, Gatter K et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res 2001; 61: 8820–8829.
Fisher SE, Scharff C . FOXP2 as a molecular window into speech and language. Trends Genet 2009; 25: 166–177.
Li S, Weidenfeld J, Morrisey EE . Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol 2004; 24: 809–822.
Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA . Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol 2003; 460: 266–279.
Teramitsu I, Kudo LC, London SE, Geschwind DH, White SA . Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J Neurosci 2004; 24: 3152–3163.
French CA, Groszer M, Preece C . Coupe AM, Rajewsky K, Fisher SE. Generation of mice with a conditional Foxp2 null allele. Genesis 2007; 45: 440–446.
Fujita E, Tanabe Y, Shiota A, Ueda M, Suwa K, Momoi MY et al. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc Natl Acad Sci USA 2008; 105: 3117–3122.
Groszer M, Keays DA, Deacon RM, de Bono JP, Prasad-Mulcare S, Gaub S et al. Impaired synaptic plasticity and motor learning in mice with a point mutation implicated in human speech deficits. Curr Biol 2008; 18: 354–362.
Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE et al. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development 2004; 131: 4477–4487.
Hu H, Wang B, Borde M, Nardone J, Maika S, Allred L et al. Foxp1 is an essential transcriptional regulator of B cell development. Nat Immunol 2006; 7: 819–826.
Dasen JS, De Camilli A, Wang B, Tucker PW, Jessell TM . Hox repertoires for motor neuron diversity and connectivity gated by a single accessory factor, FoxP1. Cell 2008; 134: 304–316.
Feng X, Ippolito GC, Tian L, Wiehagen K, Oh S, Sambandam A et al. Foxp1 is an essential transcriptional regulator for the generation of quiescent naive T cells during thymocyte development. Blood 2010; 115: 510–518.
Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 1999; 23: 99–103.
Alexander GE, Crutcher MD, DeLong MR . Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, "prefrontal" and "limbic" functions. Prog Brain Res 1990; 85: 119–146.
Koch M . The neurobiology of startle. Prog Neurobiol 1999; 59: 107–128.
Williams DL, Goldstein G, Carpenter PA, Minshew NJ . Verbal and spatial working memory in autism. J Autism Dev Disord 2005; 35: 747–756.
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011; 147: 235–246.
Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM et al. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007; 318: 71–76.
Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012; 486: 261–265.
Clark RE, Zola SM, Squire LR . Impaired recognition memory in rats after damage to the hippocampus. J Neurosci 2000; 20: 8853–8860.
Ploeger GE, Spruijt BM, Cools AR . Spatial localization in the Morris water maze in rats: acquisition is affected by intra-accumbens injections of the dopaminergic antagonist haloperidol. Behav Neurosci 1994; 108: 927–934.
Sargolini F, Roullet P, Oliverio A, Mele A . Effects of intra-accumbens focal administrations of glutamate antagonists on object recognition memory in mice. Behav Brain Res 2003; 138: 153–163.
Peca J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011; 472: 437–442.
Qiu A, Adler M, Crocetti D, Miller MI, Mostofsky SH . Basal ganglia shapes predict social, communication, and motor dysfunctions in boys with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 539–551, e1–4.
Langen M, Kas MJ, Staal WG, van Engeland H, Durston S . The neurobiology of repetitive behavior: of mice. Neurosci Biobehav Rev 2011; 35: 345–355.
Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012; 486: 256–260.
Perry W, Minassian A, Lopez B, Maron L, Lincoln A . Sensorimotor gating deficits in adults with autism. Biol Psychiatry 2007; 61: 482–486.
Rubenstein JL . Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol 2010; 23: 118–123.
Desai NS, Rutherford LC, Turrigiano GG . Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 1999; 2: 515–520.
Koon HB, Ippolito GC, Banham AH, Tucker PW . FOXP1: a potential therapeutic target in cancer. Expert Opin Ther Targets 2007; 11: 955–965.
Vernes SC, Oliver PL, Spiteri E, Lockstone HE, Puliyadi R, Taylor JM et al. Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet 2011; 7: e1002145.
Bacon C, Rappold GA . The distinct and overlapping phenotypic spectra of FOXP1 and FOXP2 in cognitive disorders. Hum Genet 2012; 131: 1687–1698.
Watkins KE, Vargha-Khadem F, Ashburner J, Passingham RE, Connelly A, Friston KJ et al. MRI analysis of an inherited speech and language disorder: structural brain abnormalities. Brain 2002; 125: 465–478.
French CA, Jin X, Campbell TG, Gerfen E, Groszer M, Fisher SE et al. An aetiological Foxp2 mutation causes aberrant striatal activity and alters plasticity during skill learning. Mol Psychiatry 2011; 17: 1077–1085.
Acknowledgements
We thank Professor Gunther Schütz for providing the Cre deleter mice and Dr Phil Tucker for the floxed Foxp1 mice. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Ra 380/15-1). Gudrun A. Rappold is a member of the CellNetworks Cluster of Excellence.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Bacon, C., Schneider, M., Le Magueresse, C. et al. Brain-specific Foxp1 deletion impairs neuronal development and causes autistic-like behaviour. Mol Psychiatry 20, 632–639 (2015). https://doi.org/10.1038/mp.2014.116
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2014.116
This article is cited by
-
Possible roles of deep cortical neurons and oligodendrocytes in the neural basis of human sociality
Anatomical Science International (2024)
-
Targeted mutagenesis in mice via an engineered AsCas12f1 system
Cellular and Molecular Life Sciences (2024)
-
Cortex-restricted deletion of Foxp1 impairs barrel formation and induces aberrant tactile responses in a mouse model of autism
Molecular Autism (2023)
-
Single-cell epigenomics and spatiotemporal transcriptomics reveal human cerebellar development
Nature Communications (2023)
-
Inhibition of Foxp4 Disrupts Cadherin-based Adhesion of Radial Glial Cells, Leading to Abnormal Differentiation and Migration of Cortical Neurons in Mice
Neuroscience Bulletin (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}