
Abstract
Myxofibrosarcomas are morphologically heterogeneous soft tissue sarcomas lacking a specific immunohistochemical expression profile and recurrent genetic changes. The study was designed to gain further insights into the molecular landscape of myxofibrosarcomas by targeted re-sequencing of known cancer driver hotspot mutations and the analysis of genomewide somatic copy number alterations. A well-defined group of myxofibrosarcomas, including myxofibrosarcomas G1 (n=6), myxofibrosarcomas G3 (n=7), myxofibrosarcomas with morphologically heterogeneous and independently selectable G1 and G3 areas within a tumor (n=8), and myxofibrosarcomas G3 with subsequent tumor recurrence (n=1) or metastatic disease (n=3) were evaluated. Mutational analysis demonstrated mutations in TP53, PTEN, FGFR3, CDKN2A, and RB1. TP53 mutations were seen in 11 (44%) of patients and detected in myxofibrosarcomas G1, G3, with heterogeneous morphology and G3 with subsequent metastases in 1 patient (16%), 3 patients (42%), 2 patients (62.5%), and 3 patients (75%), respectively. Additional mutations were detected in 2 patients, intratumoral mutational heterogeneity in 1 patient. We observed a variety of copy number alterations typical for myxofibrosarcomas, with higher numbers in G3 compared with G1 myxofibrosarcomas. Cluster analysis revealed distinctive features especially in metastatic and recurrent disease. Focal alterations affected CDKN2A, CCND1, CCNE1, EGFR, EPHA3, EPHB1, FGFR1, JUN, NF1, RB1, RET, TP53, and additional novel amplifications in CCNE1, KIT, EGFR, RET, BRAF, NTRK2 were seen in G3 compared with the G1 tumor areas. The total number of focal events in G1 versus G3 tumors differed significantly (P=0.0014). TRIO and RICTOR co-amplification was seen in 8 (44%) G3 and 1 (10%) G1 myxofibrosarcomas and RICTOR amplification alone in 4 (40%) G1 myxofibrosarcomas. TRIO amplification was significantly (P=0.0218) higher in G3 myxofibrosarcomas indicating a late genetic event. These findings support the use of expanded molecular profiling in myxofibrosarcomas to detect drug-able targets to allow patients to participate in basket trials.
Similar content being viewed by others

Main
Soft tissue sarcomas are a rare and heterogeneous group of tumors. Advances in molecular techniques provided significant insights into the biology of these tumors. The confirmation of morphological diagnoses by molecular analyses has dramatically improved standardized diagnostic approaches. The significant impact of molecular analyses on a subset of sarcomas including gastrointestinal stromal tumors, well and dedifferentiated liposarcomas and sarcomas harboring unique chromosomal translocations has recently been demonstrated.1 On a molecular point of view soft tissue sarcomas can be divided into two main groups. The first group includes sarcomas with specific genetic alterations and simple karyotypes, such as reciprocal chromosomal translocations (eg, synovial sarcoma) and specific oncogenic mutations (eg, KIT in GIST). The second group of sarcomas is characterized by non-specific genetic alterations and complex, unbalanced karyotypes as seen in leiomyosarcoma and myxofibrosarcoma.2 Myxofibrosarcoma is one of the most frequent soft tissue sarcomas in elderly patients3 and was nearly simultaneously described by two independent groups as myxoid soft tissue tumor primarily affecting the extremities.4, 5 Myxofibrosarcoma is a fibroblastic neoplasm composed of variable prominent myxoid stroma, atypical/pleomorphic tumor cells and a distinctive curvilinear vascular pattern.3 These tumors occur in superficial and deep locations and show tendency to recur in up to 50–60% of cases independent to the histological grade. Low-grade lesions may become higher-grade in subsequent recurrences and hence acquire metastatic potential.6 Despite the distinctive morphology myxofibrosarcoma lack a specific immunohistochemical expression profile, as well as a typical genetic fingerprint. Therefore, the histologic diagnosis is mainly based on HE morphology. Generally accepted biomarkers to predict recurrent disease do not exist and the molecular landscape of myxofibrosarcoma is not yet elucidated. The present study was designed to gain further insights into the molecular landscape of myxofibrosarcoma by targeted re-sequencing of known cancer driver hotspot mutations and the analysis of genome-wide somatic copy number alterations. By investigating clear cut G1 and G3 primary myxofibrosarcoma, intra-lesional heterogeneous tumor areas of primary myxofibrosarcoma, as well as comparing primary myxofibrosarcoma with subsequent recurrent myxofibrosarcoma or metastatic disease we intended to gain insights into molecular mechanisms essential for tumor-progression and at the same time to search for targetable genetic changes and biomarkers for personalized treatment strategies.
Materials and methods
Tumor Samples
To select optimal material for molecular analysis we reviewed formalin fixed and paraffin embedded myxofibrosarcoma samples from 112 patients diagnosed at the Institute of Pathology, Medical University of Graz between 1998 and 2015. For molecular analysis only tumor samples from which high-quality DNA could be extracted were selected. We selected classic cases of myxofibrosarcomas of different tumor grades including grade 1 (G1) (n=6) and grade 3 (G3) (n=7) tumors for molecular analyses. To explore intratumoral heterogeneity we selected myxofibrosarcomas that demonstrated G1 and G3 tumor areas, where manual micro-dissection could be performed without a significant risk of contamination between G1 and G3 areas (n=8). In addition we selected one case of a primary myxofibrosarcoma with intra-tumoral heterogeneity and subsequent recurrent disease (n=1), as well as three myxofibrosarcomas G3 with subsequent metastatic disease (n=3) to compare histologic features and molecular profiles of the different specimen. Clinical data are summarized in Table 1.
Four-micron hematoxylin and eosin stained sections generated from formalin-fixed paraffin embedded tissue were reviewed, independently by two pathologists (B.LA, S.S), to confirm the diagnoses before inclusion in the study (Supplementary material Table S1).
Immunohistochemistry was performed in all cases using the following antibodies and conditions: MDM2 (clone 1F2, dilution 1:50, Calbiochem), CD34 (clone QBend10, ready to use Dako Omnis), desmin (clone D33, ready to use Dako Omnis), SMA (clone 1A4, 1:5000, Sigma), S100 (polyclonal, dilution 1:2000, Dako or ready to use Dako Omnis), HMB45 (ready to use Dako Omnis), MUC4 (8G-7, 1:500, Abcam), CK (MNF116, 1:00, Dako). The Envision Plus detection system (Dako, Carpinteria, CA, USA) was used. Appropriate positive and negative controls were used throughout the investigation. Fluorescence in situ hybridization was performed by using the ZytoLight SPEC MDM2/CEN 12 Dual Color Probe (Z-2013-200, Zytovision).
Mutation Analysis by Next Generation Sequencing
For molecular analysis serial sections from 39 samples were obtained with first and last levels stained for hematoxylin and eosin to ensure optimal tissue selection. Unstained, intermediate sections were mounted on glass slides for selective manual microdissection with a scalpel. Genomic DNA was isolated on a Maxwell MDxResearch System (Promega). Using the Cancer Hotspot Panel v2 (Thermo Fisher Scientific, Waltham, MA, USA) 207 amplicons covering approximately 2,800 COSMIC mutations from 50 oncogenes and tumor suppressor genes were amplified. Amplicon libraries were prepared using the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific). Emulsion PCR and sequencing runs were performed with the appropriate kits (Ion One Touch Template Kit version 2 and Ion Proton 200 Sequencing Kit; Thermo Fisher Scientific) using Ion PI chips and sub-sequentially sequenced on an Ion Proton Sequencer (Thermo Fisher Scientific). Sequencing length was set to 520 flows and yielded reads ranging from 70 to 150 bp, consistent with the expected amplicon size range. For improved variant detection all samples were run in technical duplicates.
Next Generation Sequencing Data Analysis
Initial data analysis was performed using the Ion Torrent Suite Software version 4.1 Plug-ins (Thermo Fisher Scientific, open source, general public license). Briefly, this included base calling, alignment to the reference genome (HG19) using the Torrent Mapper and variant calling by a modified diBayes approach by taking into account the flow space information. All called variants were annotated using open source software (Annovar, http://www.openbioinformatics.org/annovar/annovardownloadform.php, and SnpEff http://snpeff.sourceforge.net/download.html; both last accessed on September 28, 2015).7, 8 Coding, nonsynonymous mutation calls present in both technical duplicates were further evaluated and visually inspected in Integrative Genomics Viewer. Variant calls resulting from technical read errors or sequence effects were excluded from the analysis.
Low-pass Whole Genome Sequencing for Copy Number Analysis
Next generation sequencing libraries were prepared using 500 ng genomic DNA and the Ion Plus Fragment Library Kit (Cat. No. 4471252, Thermo Fisher Scientific). DNA libraries were sequenced on the Ion Proton Sequencer yielding 8–10 million reads per library. Copy number analysis was performed as previously described.9 Briefly, low-coverage whole-genome sequencing reads were mapped to the pseudo-autosomal-region-masked genome and reads in different windows were counted and normalized by the total amount of reads. We further normalized read counts according to the GC-content using LOWESS-statistics. To avoid position effects we normalized the sequencing data with GC-normalized read counts of a set of nine non-neoplastic reference samples, which were prepared and sequenced the same way. Subsequently we generated segments of similar copy-number values by applying circular binary segmentation and Gain and Loss Analysis of DNA. Focal events were identified from the segmented data using previously published criteria.10 Segmented log2ratios were plotted in a heat map.
Statistics
In order to visualize copy number alterations we plotted a heat map in R. To identify overlapping copy number alterations within each sample group, we subjected our data to the Genomic Recurrent Event Viewer (GREVE) algorithm.10 The GREVE output was plotted using R circos software package (http://circos.ca/).11 Segmented log2ratios for each bin were clustered hierarchically by the hclust function provided by fastcluster package of R, using average linkage on a Manhattan distance matrix in order to identify phylogenetic relationships between copy number profiles of tumors/tumor areas from the same patient.12 Details on settings of the software parameters are available on request. Comparison of the percentage of aberrant genomic regions for each subgroup was performed in R using the two-sided fisher.test and the non-parametric wilcox.test function.12
Results
Clinical Features
Clinical data of patients and respective treatments are summarized in Table 1. The median age at presentation was 65 years (ranging from 29 to 94 years) with the majority of patients being female (18 female and 7 male patients, female to male ratio of 2.6 to 1). The size of the primary tumor ranged from 3 to 20 cm (median size 10 cm). Tumors were located superficial (n=8), deep (n=13), or superficial and deep (n=3). Six patients suffered from myxofibrosarcoma G1 and seven patients from myxofibrosarcoma G3. In eight patients the tumors were morphologically heterogeneous and demonstrated both G1 and G3 tumor areas. Three out of twenty-five patients developed a tumor recurrence, and seven patients developed metastases. In four patients the primary G3 myxofibrosarcoma, as well as the recurrent (n=1) or metastatic tumor (n=3) could be evaluated. Four patients died of the disease. The median follow-up time for the remaining patients ranged between 9 and 160 months (median 66 months).
Macroscopic and Microscopic Features
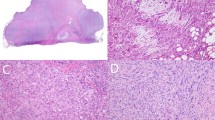
On gross examination tumors were usually multinodular, fairly well circumscribed and lacked an obvious infiltrative growth pattern (Figure 1a). The amount of myxoid tumor areas was variable. All cases showed a classic morphology with at least one tumor block demonstrating myxoid tumor areas with detectable curvilinear blood vessels (Figure 1b). The myxoid tumor component encountered at least 10% in G3 and 75% in G1 tumors. For the study a three-tier grading system was used according to the recent WHO classification of tumors of soft tissue and bone.3 Low-grade tumors/tumor areas (G1) were hypocellular with abundant myxoid matrix showing scattered atypical cells and prominent curvilinear vessels (Figure 1c).

(a) Myxofibrosarcoma in superficial location with multinodular growth pattern and abundant myxoid matrix (pt.3). (b) Scanning magnification of one slide of a myxofibrosarcoma in superficial location with multinodular and infiltrative growth pattern and partly myxoid matrix. Inset Figure 1b: Typical myxoid tumor areas with curvilinear vessels. (c) Low grade myxofibrosarcoma with abundant myxoid matrix and curvilinear blood vessels and atypical spindle cells (pt.4). (d) High power view of a high grade myxofibrosarcoma. Note the curvilinear vessel in the center (pt.9). (e) myxofibrosarcoma with abrupt transition between low and high-grade areas (primary tumor pt.22). (f) Recurrent myxofibrosarcoma from pt. 22 (left) and lung metastasis of a myxofibrosarcoma from patients 24 (right).
High-grade tumors/ tumor areas (G3) were hypercellular with solid sheets of tumor cells with moderate to severe nuclear atypia, more prominent fibrous stroma, as well as necrosis (Figure 1d). To explore myxofibrosarcomas with intratumoral heterogeneity we selected tumors with abrupt transition between low and high-grade tumor areas (Figure 1e) or tumors were low and high grade areas were seen in different tumor blocks. Tumor recurrence and metastasis (Figure 1f) showed morphologic features characteristic for myxofibrosarcoma. For detailed description of histologic features including histologic margins, cell type, cytologic atypia, necrosis, vascular invasion see Supplementary material Table S1).
Immunohistochemistry revealed a consistent lack of expression for cytokeratin, S100, HMB45, and desmin in all cases. All cases demonstrated at least a focal staining for SMA. CD34 was focally expressed in 5 out of 15 cases tested. MUC4 was negative in all 6 G1 myxofibrosarcomas. Single cells with MDM2 nuclear expression could be demonstrated in 3 out of 10 tested cases. However, none of the cases demonstrated MDM2 amplification by FISH.
Analysis of Somatic Hotspot Mutations
Hotspot mutation analysis was performed on 37 samples obtained from 25 patients. After variant prioritization (variant had to be present in both technical duplicates with a mutant allele frequency >5%, variant had to be present in less than 0.1% of the general population) we ended up with 27 putative, somatic mutations in 11 patients (44%) (Table 2). Interestingly, all 11 patients harbored TP53 mutations in their tumors, whereas two patients showed two mutations in the TP53 gene. In patients with G1 and G3 myxofibrosarcoma (lacking obvious intra-lesional tumor-heterogeneity) on histology, TP53 mutations were detected in only one (16%) and three patients (42%), respectively. In contrast, TP53 mutations could be detected in five (62.5%) patients with myxofibrosarcoma where heterogeneous G1 and G3 tumor areas could be separately sampled. Two of three patients (75%) with myxofibrosarcoma G3 and subsequent metastasis showed TP53 mutations. Moreover, in two patients (P11 and P20) additional mutations in CDKN1A, FGFR3, PTEN, and RB1 were observed.
Except for one patient (P20), in which an additional mutation in CDKN2 A was present in the G3 area the identical mutations were identified in G1 and G3 areas. In addition to somatic mutations we observed putative germline variants in ATM, JAK, KDR, KIT, MET, and PIK3CA (data not shown). The most commonly observed variant in 14 patients (56%) was Q472H in the KDR-gene. After sequencing adjacent normal tissues from our samples, the Q472H variant was detected in all samples confirming the constitutional origin. Despite the frequent presence of the variant in our patient cohort the allele frequency did not significantly differ from the general population.
Establishment of Genome-Wide Somatic Copy Number Alterations
In order to assess somatic copy number alterations we employed low-coverage whole genome sequencing. Detailed copy number profiles for each sample are shown in Figures 2a and b.

Genome-wide somatic copy number alterations established from low-coverage whole genome sequencing. Heat map depicting segmented log2-ratios of (a) low grade myxofibrosarcoma (G1) and (b) high grade myxofibrosarcoma (G3). Blue indicates loss of chromosomal material, red indicates gain of chromosomal material. (c) Circos plot showing the relative abundance of somatic copy number alterations calculated with the GREVE algorithm of myxofibrosarcoma from the TCGA (inner circle), G1 myxofibrosarcoma (middle circle) and G3 myxofibrosarcoma (outer circle). Outer ideogram runs clockwise from chromosome 1 to chromosome X with labels in Mb of physical distance.
To identify recurrent somatic copy number alteration and to investigate whether somatic copy number alterations differ in G1 and G3 myxofibrosarcoma, in morphologic heterogeneous areas (G1 and G3) within a myxofibrosarcoma or between primary myxofibrosarcoma and subsequent tumor recurrence/metastases, we subjected our data to the GREVE algorithm (Figure 2c). In general, we observed a higher amount of somatic copy number alterations in G3 tumors compared with G1 tumors. When we compared the copy number profiles to 22 publicly available datasets from myxofibrosarcomas from The Cancer Genome Atlas we observed marked similarities (Figure 2c) such as loss of chromosome arms and chromosomes 1p and 13 and gains of 5p or 9p. To test whether morphologically different tumor areas of a myxofibrosarcoma or tumors (primary myxofibrosarcoma/recurrence/metastasis) from the same patients are phylogenetically related we performed hierarchical clustering (Figure 3a). With the exception of samples from patients 14 and 18, all samples from the same tumor clustered together in the same branch confirming the lack of heterogeneity observed from the mutation analysis and suggesting a common origin. Likewise, the G1 area and the recurrence sample from patient 22 branched together, while the G3 tumor formed its own branch. Similarly, the, metastatic tumor samples (P24, P25) appeared to be genetically different from their respective primary tumors.

(a) Hierarchical cluster analysis (Manhattan distances of copy number profiles) of G1, G3 myxofibrosarcoma, as well as two metastases and one recurrent tumor from 11 patients. Tumor samples from the same patients appear in the same color. With a few exceptions tumors from the same patients are located in the same branch. (b) Box plots showing median and quartiles of focal amplification of G1 and G3 tumors. The whisker caps of the box plots show the mean 5th and 95th percentile values. A Wilcoxon test revealed a significant difference of the total number of focal amplification.
When we applied our recently published algorithm for focal amplification/deletion calling13 we identified a variety of known cancer driver genes such as CDKN2A, CCND1, CCNE1, EGFR, EPHA3, EPHB1, FGFR1, JUN, NF1, RB1, RET, and TP53 (Table 3). When we compared the total number of focal events in all G1 tumors versus all G3 tumors we observed a significantly higher number in G3 tumors (P=0.0014, Figure 3b). Despite a relative homogenous profile within the same patient with respect to genome-wide somatic copy number alteration, G3 tumor areas of the same tumor showed novel emerged focal amplifications including CCNE1, KIT, EGFR, RET, BRAF, NTRK2 compared with the respective G1 areas. Two exemplary cases are displayed in Figure 4. In patient 14 focal amplifications harboring RET and CCND1 were identified which were not present in the G1 samples. A similar finding was identified in patient 19 where a novel RET amplification emerged in the G3 area of the tumor.

Genome-wide copy number profiles of tumors from (a) patients 14 and (b) patients 19. The upper panel shows the G1 tumor areas, the middle panel shows the G3 areas. The lower panel represents a differential plot of G1 and G3 indicating the emergence of novel focal amplifications. The X-axes denote chromosomes 1-22 and the gonosomes. The y-axes show log2-ratios. Gains (log2-ratio >0.2) are shown in red and losses (log2-ratio <0.2) are shown in blue. Green indicates balanced regions.
To check for the recently reported co-amplification of TRIO and RICTOR,14 we applied a log2-ratio threshold of 0.3 for the presence of a gain on chromosome arm 5p. 8/18 of G3 tumors (44%) showed a co-amplification of TRIO and RICTOR, while the same phenomenon was observed in only one G1 tumor (10%, not significant). In contrast, overrepresentation of RICTOR alone was found in 4 out of 10 of G1 tumors (40%) indicating that TRIO amplification is significantly more abundant in G3 tumors (P=0.022). Focal amplifications harboring either RICTOR or TRIO were observed in a total of five samples (RICTOR: 2 × G3 and 1 × G1; TRIO: 2 × G3).
Discussion
Over the last two decades advances in molecular classification have led to increased reproducibility/consistency of diagnoses of soft tissue tumors among pathologists. Moreover, genetic profiling has enabled guidance for treatment decisions and can be used for patient stratification based on genetic changes.15, 16, 17, 18 Systemic treatment of patients suffering from MFS has not changed over the years based on the highly complex genetic changes and the lack of recurrent characteristic molecular alterations in myxofibrosarcomas.19, 20, 21
Herein we performed molecular profiling in myxofibrosarcomas to unveil clinically actionable targets and to gain further insights in mechanisms involved in tumor progression. Although myxofibrosarcomas are genetically highly complex sarcomas, the abundance of somatic mutations was low. Somatic mutations were identified in only 11 (44%) patients. Of note, all these patients showed at least one somatic TP53 mutation. Additional mutations were only found in two patients. Patient 11, suffering from a G3 myxofibrosarcoma showed a mutation in FGFR3. In patient 20 mutations in PTEN and RB1 were detected in both G1 and G3 tumor areas, while a novel mutation in CDKN2A emerged in the G3 area. The presence of identical TP53 mutations in morphologically heterogeneous (G1 and G3) tumor areas within a primary myxofibrosarcoma was seen in 5 (62.5%) patients. Interestingly, TP53 mutations were only detected in one patient (16%) with pure G1 myxofibrosarcoma and 3 patients (42%) with pure G3 M myxofibrosarcoma. The crucial role of TP53 mutations has been previously shown in mouse models demonstrating that KRAS and TP53 mutations are commonly involved in sarcomagenesis.22 In contrast, in a study by Willems et al. no mutations in KRAS codon 12/13 or in TP53 could be detected in 10 G1 myxofibrosarcomas.23 Nevertheless, mutations in the TP53 gene have been commonly reported in both mesenchymal and solid tumor entities and have been linked to poor prognosis and chemotherapy-resistance.24 Moreover, it was demonstrated that TP53 mutations are relatively common in sarcomas with non-specific genetic aberrations and complex karyotypes compared to sarcomas with reciprocal specific translocations.25 In general, two types of mutations affecting TP53 have been reported (i) loss of function (LOF) mutations, which adhere to the two-hit hypothesis by two inactivating mutation or a mutation in one allele and simultaneous deletions in regions of the 17p chromosome encompassing the TP53 locus and (ii) gain of function mutations, which mostly act in a dominant negative manner.26 In our cohort both types of mutation were identified though without a clear association regarding tumor grade. These data suggest that TP53 mutations might be an early event and act as a driver for tumor progression in a subset of myxofibrosarcomas.
Because there is evidence that germline variants may have an impact on tumor progression and clinical outcome,27, 28 we assessed the presence of nonsynonymous germline variants.29
Because of the putative functional relevance of the KDR Q472H variant,30, 31 we checked whether the variant differed from the Hardy–Weinberg equilibrium for expected frequencies of each genotype. Despite the frequent presence of the variant in our patient cohort the allele frequency did not significantly differ from the general population suggesting a limited impact on oncogenesis.32, 33 However, the KDR (kinase insert domain receptor, also known as VEGFR2) Q472H variant has been associated with an angiogenic phenotype characterized by increased tumor vascular density and VEGF secretion. Studies on melanoma cells harboring KDR Q472H demonstrated higher proliferative and invasive capacity and sensitivity to targeted inhibition of VGFR2.27 Similar antiangiogenic approaches have been discussed for other tumor31, 34, 35, 36, 37 although the role of the KDR germline variant is still controversially discussed in literature. Nevertheless, the recently published MUG-Myx2 cell lines might help to gain further insights if antiangiogenic treatment approaches might be a potential additional treatment option for a subset of patients suffering from myxofibrosarcoma.38
A genome-wide assessment of copy number alterations revealed similar profiles as reported datasets from G3 myxofibrosarcomas (n=22) from The Cancer Genome Atlas. As expected G3 tumors showed a higher amount of somatic copy number alterations than G1 tumors. This is consistent with data from other soft tissue sarcomas, in which significantly increased numbers of somatic copy number alteration were reported in leiomyosarcoma.39 These data confirm prior studies demonstrating that high grade myxofibrosarcoma is among the most highly complex sarcoma types.19, 20, 21 Hierarchical clustering based on copy number alterations revealed — except for two patients — a phylogenetic relationship of tumors/tumor areas from the same patient. It is of note that the G1 tumor of patient 18 showed a balanced profile most likely due to low tumor content in the sample resulting in an informative result. Although G1 and G3 areas of P14 share many somatic copy number alterations, there are marked differences on chromosome 1, which might be a possible explanation for the separation of G1 and G3 as more weight is assigned to larger chromosomes during clustering. The same might apply for the metastases of P25 and P24. Although two independent metastatic samples show highly constituent profiles and cluster together in the same branch, they are clearly separated from the primary tumor, despite an obvious common origin. However, it is striking that the metastatic samples have a less complex karyotype than it was observed in the primary sample. The same phenomenon, even if less evident due to low tumor content in the metastatic sample, was observed for P25. Similar data were shown for leiomyosarcoma metastases,39 as well as for breast cancer, where regional lymph node metastases had a less complex composition compared with their primary indicating that in primary tumors and metastatic cell clones diverge early on during tumor progression and evolve independently.40
As focal events are meant to be drivers for tumorigenesis and progression we applied our recently published algorithms for the detection of focal events.13 Although focal events such as amplifications of EPHA3, FGFR1, CCNE1 or losses of NF1, CDKN2A, RB1 were identified in G1 tumors/tumor areas, G3 tumor/tumor areas showed significantly increased numbers of focal events. Interestingly, despite a relative homogenous profile with respect to genome-wide somatic copy number alterations, G3 tumor areas of the same tumor showed novel emerged focal amplifications compared to the G1 areas. These data confirm the previously reported deregulation of pathways through extensive loss of known tumor suppressors such as CDKN2A, RB1, TP53, in combination with NF1 and PTEN mutations.19 Due to the presence of NF1 mutations in 10.5% of myxofibrosarcomas the use of mTOR inhibitors has been suggested 19.41 Since we used a hotspot panel for mutation analysis, which did not cover NF1, we were unable to inform about the NF1 mutation status. However, loss of NF1 was observed in two patients. Moreover, a variety of actionable focal amplifications could be identified and might offer a more targeted driven treatment approach for patients with progressive disease. Referring to the Drug-Gene-Interaction Database42 a total of 27 focal events were potentially drug-able, which include genes that currently may not yet be targeted therapeutically but are potentially drug-able.42 Many of those, such as BRAF, EGFR, FGFR, KIT or RET, are indeed actionable targets and are actively being used for precision medicine in different tumor entities.
Recently Okada et al14 demonstrated through genomic analysis, knockdown and biochemical studies that TRIO and RICTOR play a crucial role in myxofibrosarcoma pathogenesis. With our patient cohort we obtained comparable results since gains of chromosome 5p were frequently detected. In contrast to previous studies we focused in addition to G3 myxofibrosarcomas on G1 myxofibrosarcomas and evaluated the co-amplification of TRIO and RICTOR. Thereby we made an interesting observation: while 44% of G3 tumors showed a co-amplification of TRIO and RICTOR, the same phenomenon was observed in only one G1 tumor (10%). However, overrepresentation of RICTOR alone was found in 4/10 (40%) of G1 tumors indicating that TRIO amplification is significantly more abundant in G3 tumors (P=0.0218) and therefore a late event. Our data together with the data from Okada et al. demonstrate a high dependency of myxofibrosarcoma cells on the integrin-alpha10/TRIO/RICTOR signaling and propose this signaling pathway as promising therapeutic target.
Conclusion
In summary our data suggest that patients suffering from advanced myxofibrosarcoma might benefit from expanded molecular evaluation to detect drug-able targets. As next generation sequencing technologies have become increasingly economical in the last years,43 genetic profiling on a genome-wide and gene-specific level is about to be translated into oncology practice. New clinical trial schemes, such as ‘basket trials’, are accrued to treat patients based on specific targetable genetic alterations independent of tumor histiotype44 and the significance of classifying tumors based on defining genetic alterations might be particularly relevant for myxofibrosarcomas patients.
References
Neuville A, Ranchere-Vince D, Dei Tos AP et al. Impact of molecular analysis on the final sarcoma diagnosis: a study on 763 cases collected during a European epidemiological study. Am J Surg Pathol 2013;37:1259–1268.
Antonescu CR . The role of genetic testing in soft tissue sarcoma. Histopathology 2006;48:13–21.
Fletcher CDM World Health Organization, International Agency for Research on Cancer WHO Classification of Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2013.
Weiss SW, Enzinger FM . Myxoid variant of malignant fibrous histiocytoma. Cancer 1977;39:1672–1685.
Angervall L, Kindblom LG, Merck C . Myxofibrosarcoma. A study of 30 cases. Acta Pathol Microbiol Scand A 1977;85A:127–140.
Mentzel T, Calonje E, Wadden C et al. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol 1996;20:391–405.
Cingolani P, Platts A, Wang le L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012;6:80–92.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
Heitzer E, Ulz P, Belic J et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome Med 2013;5:30.
Cazier JB, Holmes CC, Broxholme J . GREVE: Genomic Recurrent Event ViEwer to assist the identification of patterns across individual cancer samples. Bioinformatics 2012;28:2981–2982.
Krzywinski M, Schein J, Birol I et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009;19:1639–1645.
RDC T . A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2008.
Ulz P, Belic J, Graf R et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Commun 2016;7:12008.
Okada T, Lee AY, Qin LX et al. Integrin-alpha10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov 2016;6:1148–1165.
Group ESESNW. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 (Suppl 3):iii102–iii112.
Group ESESNW. Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25 (Suppl 3):iii21–iii26.
von Mehren M, Randall RL, Benjamin RS et al. Gastrointestinal stromal tumors, version 2.2014. J Natl Compr Canc Netw 2014;12:853–862.
von Mehren M, Randall RL, Benjamin RS et al. Soft tissue sarcoma, version 2.2014. J Natl Compr Canc Netw 2014;12:473–483.
Barretina J, Taylor BS, Banerji S et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet 2010;42:715–721.
Gibault L, Perot G, Chibon F et al. New insights in sarcoma oncogenesis: a comprehensive analysis of a large series of 160 soft tissue sarcomas with complex genomics. J Pathol 2011;223:64–71.
Taylor BS, Barretina J, Maki RG et al. Advances in sarcoma genomics and new therapeutic targets. Nat Rev Cancer 2011;11:541–557.
Kirsch DG, Dinulescu DM, Miller JB et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med 2007;13:992–997.
Willems SM, van Remoortere A, van Zeijl R et al. Imaging mass spectrometry of myxoid sarcomas identifies proteins and lipids specific to tumour type and grade, and reveals biochemical intratumour heterogeneity. J Pathol 2010;222:400–409.
Rivlin N, Brosh R, Oren M et al. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2011;2:466–474.
Borden EC, Baker LH, Bell RS et al. Soft tissue sarcomas of adults: state of the translational science. Clin Cancer Res 2003;9:1941–1956.
Miller M, Shirole N, Tian R et al. The evolution of TP53 mutations: from loss-of-function to separation-of-function mutants. J Cancer Biol Res 2016;4:4.
Silva IP, Salhi A, Giles KM et al. Identification of a novel pathogenic germline KDR variant in melanoma. Clin Cancer Res 2015;22:2377–2385.
Rendleman J, Shang S, Dominianni C et al. Melanoma risk loci as determinants of melanoma recurrence and survival. J Transl Med 2013;11:279.
Jones S, Anagnostou V, Lytle K et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 2015;7:283ra253.
Lee SY, Haq F, Kim D et al. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS ONE 2014;9:e90459.
Glubb DM, Cerri E, Giese A et al. Novel functional germline variants in the VEGF receptor 2 gene and their effect on gene expression and microvessel density in lung cancer. Clin Cancer Res 2011;17:5257–5267.
Gao Y, Ma P, He Y et al. Genetic variations of kinase inserts domain receptor (KDR) gene are associated with the risk of astrocytomas. Mol Neurobiol 2016;53:2541–2549.
Ray-Coquard I, Italiano A, Bompas E et al. Sorafenib for patients with advanced angiosarcoma: a phase II Trial from the French Sarcoma Group (GSF/GETO). Oncologist 2012;17:260–266.
Masago K, Fujita S, Muraki M et al. Next-generation sequencing of tyrosine kinase inhibitor-resistant non-small-cell lung cancers in patients harboring epidermal growth factor-activating mutations. BMC Cancer 2015;15:908.
Dong G, Guo X, Fu X et al. Potentially functional genetic variants in KDR gene as prognostic markers in patients with resected colorectal cancer. Cancer Sci 2012;103:561–568.
Hansen TF, Sorensen FB, Spindler KL et al. Microvessel density and the association with single nucleotide polymorphisms of the vascular endothelial growth factor receptor 2 in patients with colorectal cancer. Virchows Arch 2010;456:251–260.
Loaiza-Bonilla A, Jensen CE, Shroff S et al. KDR mutation as a novel predictive biomarker of exceptional response to regorafenib in metastatic colorectal cancer. Cureus 2016;8:e478.
Lohberger B, Stuendl N, Leithner A et al. Establishment of a novel cellular model for myxofibrosarcoma heterogeneity. Sci Rep 2017;7:44700.
Liegl-Atzwanger B, Heitzer E, Flicker K et al. Exploring chromosomal abnormalities and genetic changes in uterine smooth muscle tumors. Mod Pathol 2016;29:1262–1277.
Ellsworth RE, Ellsworth DL, Neatrour DM et al. Allelic imbalance in primary breast carcinomas and metastatic tumors of the axillary lymph nodes. Mol Cancer Res 2005;3:71–77.
Johannessen CM, Reczek EE, James MF et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA 2005;102:8573–8578.
Wagner AH, Coffman AC, Ainscough BJ et al. DGIdb 2.0: mining clinically relevant drug-gene interactions. Nucleic Acids Res 2016;44:D1036–D1044.
Van Allen EM, Wagle N, Stojanov P et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med 2014;20:682–688.
Redig AJ, Janne PA . Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol 2015;33:975–977.
Acknowledgements
We would like to thank Andrea Thüringer, Iris Halbwedl, Margit Gogg-Kamerer, Sylvia Eidenhammer, and Fritz Jammnig for their technical assistance. The study was funded by the ÖNB Jubiläumsfond (1619).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Heitzer, E., Sunitsch, S., Gilg, M. et al. Expanded molecular profiling of myxofibrosarcoma reveals potentially actionable targets. Mod Pathol 30, 1698–1709 (2017). https://doi.org/10.1038/modpathol.2017.94
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.94
This article is cited by
-
A novel patient-derived immortalised cell line of myxofibrosarcoma: a tool for preclinical drugs testing and the generation of near-patient models
BMC Cancer (2023)
-
Molecular and clinicopathological analysis revealed an immuno-checkpoint inhibitor as a potential therapeutic target in a subset of high-grade myxofibrosarcoma
Virchows Archiv (2022)
-
T-regulatory cells predict clinical outcome in soft tissue sarcoma patients: a clinico-pathological study
British Journal of Cancer (2021)
-
Immunohistochemical selection of biomarkers for tumor-targeted image-guided surgery of myxofibrosarcoma
Scientific Reports (2020)
-
Fibroblast growth factor receptors as treatment targets in clinical oncology
Nature Reviews Clinical Oncology (2019)
