
Abstract
The amplification of chromosome 9p24.1 encoding PD-L1, PD-L2, and JAK2 has been reported in multiple types of cancer and is associated with poor outcome, upregulation of PD-L1, and activation of the JAK/STAT pathway. We have developed a novel fluorescence in situ hybridization assay which combines 3 probes mapping to 9p24.1 with a commercial chromosome 9 centromere (CEN9) probe for detection of the JAK2/9p24.1 amplification. JAK2 fluorescence in situ hybridization was compared with array-based comparative genomic hybridization in 34 samples of triple negative breast cancer tumor. By array-based comparative genomic hybridization, 15 had 9p24.1 copy-number gain (log2ratio>0.3) and 19 were classified as non-gain (log2ratio≤0.3). Copy-number gain was defined as JAK2/CEN9 ratio ≥1.1 or average JAK2 signals≥3.0. Twelve of 15 samples with copy-number gain by array-based comparative genomic hybridization were also detected by fluorescence in situ hybridization. Eighteen of 19 samples classified as copy-number non-gain by array-based comparative genomic hybridization were concordant by array-based comparative genomic hybridization. The sensitivity and specificity of the fluorescence in situ hybridization assay was 80% and 95%, respectively (P=0.02). The sample with the highest level of amplification by array-based comparative genomic hybridization (log2ratio=3.6) also scored highest by fluorescence in situ hybridization (ratio=8.2). There was a correlation between the expression of JAK2 and amplification status (Mean 633 vs 393, P=0.02), and there was a trend of association with PD-L1 RNA expression (Mean 46 vs 22, P=0.11). No significant association was observed between PD-L1 immunohistochemistry expression and copy-number gain status. In summary, the novel array-based comparative genomic hybridization assay for detection of chromosome 9p24.1 strongly correlates with the detection of copy-number gain by array-based comparative genomic hybridization. In triple negative breast cancer, this biomarker may identify a relevant subset of patients for targeted molecular therapies.
Similar content being viewed by others


Main
Chromosome 9p24.1 amplification has been detected in lymphoma, non-small cell lung cancer, colorectal cancer, and in the subset of triple-negative breast cancers that lack overexpression of receptors for estrogen, progesterone, and the human epidermal growth factor 2 (HER2).1, 2, 3 The shortest overlapping region of amplification includes the genes encoding JAK2, PD-L1, and PD-L2, and is associated with aggressive behavior and poor outcome.2, 4, 5 JAK2 contributes to pathways involved in promoting cell proliferation and metastasis,6 and PD-L1 and PD-L2 contribute to immune evasion.7
Amplification of chromosome 9p24.1 upregulates JAK2 expression and activates the JAK2/STAT3 pathway,1, 2 and increases PD-L1 mRNA expression in triple negative breast cancer.2 Approximately 20% of triple negative breast cancer have overexpression of PD-L1 protein in tumor cells which may lead to immune escape.8 A study in classical Hodgkin’s lymphoma (cHL) found that the 9p24.1 alterations are a defining feature5 and correlate with responsiveness to checkpoint inhibitors.9 These data suggest that 9p24.1 amplification could be a clinically relevant predictive biomarker for response to JAK2 inhibition and/or PD-1/PD-L1 checkpoint blockade. A subset of triple negative breast cancer patients benefit from targeted blockade of PD-1/PD-L1,10 although predictive biomarkers of response have not been identified.11, 12 In lymphoma and myelofibrosis, JAK2 inhibitors have evidence of clinical efficacy,1, 13 however in breast cancer, JAK2 inhibition remains in early-phase clinical trials.14
For both JAK2 inhibitor and PD-1/PD-L1 blockade clinical trials, receptor overexpression has been discordant with response to therapy.10, 15 In a study of 296 patients with multiple tumor types including melanoma, lung cancer, and colorectal cancer treated with nivolumab, only 36% of PD-L1 immunohistochemistry positive tumors responded to nivolumab, demonstrating the need for improved patient selection.9 In addition, PD-L1 is rapidly inducible by interferon-γ (IFN-γ), which is expressed by both tumor cells and tumor-infiltrating lymphocytes and is involved in maintaining a tumor suppressive immune microenvironment.16 The regulation of PD-L1 expression is complex and not fully understood. Chromosome 9p24.1 amplification was found to upregulate PD-L1 expression in cHL1 but not in breast cancer in vitro (unpublished observations). Activation of mTOR-AKT and PTEN loss have been reported to trigger the expression of PD-L1 in lung cancer17 and breast cancer.18 The expression of PD-L1 is not only dependent on DNA amplification but also modulated by the tumor immune microenvironment.19 Quantitation RNA profiling has been used to identify immune-related gene signatures in breast cancer that correlate with pathologic complete response and survival in neoadjuvant chemotherapy treated triple negative breast cancer patients.20 These studies demonstrate that biomarkers of the tumor immune microenvironment may identify additional targets for triple negative breast cancer therapy.
Array-based comparative genomic hybridization is emerging as a standard benchmark assay for detection and quantitation copy-number variation, but the application of array-based comparative genomic hybridization for widespread clinical use is limited by yield, throughput and sample quality.21, 22, 23 Targeted fluorescent in situ hybridization has been standardized for clinical detection of copy-number aberrations,24, 25 such as for HER-2/neu.23 To distinguish true HER-2/neu amplification from chromosome 17 polysomy, the HER-2 to chromosome 17 ratio is used to define amplification status, with a 97% concordance between array-based comparative genomic hybridization and fluorescence in situ hybridization reported.23 fluorescence in situ hybridization targeting 9p24.1, including either PD-L1 or JAK2, has been evaluated in tissue from cHL patients, however for breast cancer it has only been applied in animal models and cell lines.4, 5 PD-L1 is tightly linked with JAK2 (<400 kb), so that development of a fluorescence in situ hybridization probe for JAK2 will likely be a surrogate for PD-L1 copy-number evaluation.
In this study, we developed and evaluated a clinically applicable fluorescence in situ hybridization assay to detect JAK2/9p24.1 copy-number alterations. We directly compare 9p24.1 copy-number and CEN9 copy-number with array-based comparative genomic hybridization quantitation from flow cytometry-sorted tumor nuclei derived from tumor tissue. We determined the association of 9p24.1 copy-number alteration with JAK2 and PD-L1 mRNA expression.
Materials and methods
Tissue Samples
A total of 40 formalin-fixed paraffin-embedded primary triple negative breast cancer tissue surgical pathology samples were obtained from Mayo Clinic Arizona. Estrogen receptor and progesterone receptor expression were determined using central pathologic review. HER-2/neu expression was determined by immunohistochemistry and graded by immunohistochemistry+/−fluorescence in situ hybridization under CLIA/CAP guidelines. Of these, 38 samples had available array-based comparative genomic hybridization data from prior research study2 and were included for JAK2/9p24.1/fluorescence in situ hybridization analysis. We also evaluated 17 of these samples for RNA expression using the Nanostring nCounter PanCancer Immune Profiling Panel (nanoString Technologies, Seattle, WA). Medical records were reviewed for clinical information and pathology reports. All patients provided written informed consent for the use of tissue samples and medical records and the study was approved by the Mayo Clinic Institutional Review Board. The workflow of sample selection at each step is illustrated in Figure 1.

Work flow chart of tumor samples included in the study. A total of 40 formalin-fixed paraffin-embedded primary triple negative breast cancer tissues were analyzed by array-based comparative genomic hybridization(aCGH). Of these, 38 tissue samples were eligible for evaluation by fluorescence in situ hybridization (FISH) assay and 17 samples were further ran on the Nanostring nCounter platform. DCIS: Ductal carcinoma in situ.
Microarray-Based Comparative Genomic Hybridization Analysis
Tumor nuclei were stained with DAPI, and the aneuploidy fraction was selected by flow cytometry. DNA from flow cytometry-sorted nuclei was extracted using an amended protocol from QIAamp DNA Micro Kit from Qiagen (Valencia, CA), which has been described previously.2 DNA was prepared for CGH arrays (Agilent Technologies, Santa Clara, CA) and array-based comparative genomic hybridization data were analyzed using standardized protocols.2 Briefly, all microarray slides were scanned by an Agilent 2565C DNA scanner and the images were analyzed with Agilent Feature Extraction version 10.7 using default settings. The array-based comparative genomic hybridization data was assessed with a series of QC metrics then analyzed using an aberration detection algorithm (ADM2)26 to define and rank 9p24.1 copy-number alterations; copy-number gain was defined as array-based comparative genomic hybridization log2 ratio >0.3.
Fluorescence In Situ Hybridization Analysis
We developed a fluorescence in situ hybridization probe mapping to 9p24.1 with home-brew JAK2 DNA (clones RP11-980L14, RP11-927H16, and CTD-2506A8)27 labeled with SpectrumOrange dUTP per manufacturer’s instructions (Abbott Molecular/fluorescence in situ hybridization Vysis Products, Abbott Park, IL). A commercially available chromosome 9 centromere (CEN9) probe with SpectrumGreen (Abbott Molecular) was combined with the JAK2/9p24.1 probes as one probe set. The enumeration probe set was applied to individual slides, hybridized,, and washed according to the Mayo Cytogenetics Core Lab partially automatic protocol.28, 29 fluorescence in situ hybridization was performed in 38 formalin-fixed paraffin-embedded tumor samples, 4 samples were excluded from analysis due to minimal tumor embedded in samples (n=3) or two populations present in the sample (n=1). For each sample, a total of 100 individual nuclei were manually scored with a fluorescence microscope, 50 nuclei by each experienced pathologist. The ratio of JAK2 average signals to CEN9 average signals was used to determine copy-number alteration of JAK2/9p24.1. Copy-number gain was defined as ratio ≥1.1 or average JAK2 signals≥3.0.4, 5, 30
Immunohistochemistry
Immunohistochemistry for PD-L1 was performed by Merck Research Laboratories (Merck & Co, Inc, Kenilworth, NJ) as in the previous study.31 Briefly, whole tissue sections cut from formalin-fixed paraffin-embedded tissue blocks were deparaffinized and subjected to heat induced epitope retrieval in Envision FLEX Target Retrieval Solution (Dako, Carpineteria CA). Endogenous peroxidase in tissues was blocked by incubation of slides in 3% hydrogen peroxide solution prior to incubation with the anti-PD-L1 antibody (clone 22C3, Merck Research Laboratories, Palo Alto, CA) for 60 min. Antigen-antibody binding was visualized with DAB chromogen (Dako, Carpineteria, CA,) and counterstained with hematoxylin. We used an established scoring system to report the PD-L1 expression level in each sample.31 Scoring of PD-L1 was conducted by a pathologist blinded to patient characteristics and clinical outcomes. A semi-quantitative 0–5 scoring system was applied; Negative: 0, Rare: 1=individuated positive cells or only very small focus within or directly adjacent to tumor tissue, Low: 2=infrequent small clusters of positive cells within or directly adjacent to tumor tissue, Moderate: 3=single large cluster, multiple smaller clusters, or moderately dense diffuse infiltration, within or directly adjacent to tumor tissue, High: 4=single very large dense cluster, multiple large clusters, or dense diffuse infiltration, Very high: 5 Coalescing clusters, dense infiltration throughout the tumor tissue. Evaluations were relativized to the size of the tumor sample. A positive PD-L1 immunohistochemistry was defined as a PD-L1 score ≥3.
RNA Expression Analysis
Total RNA was extracted from formalin-fixed paraffin-embedded sections using RNeasy formalin-fixed paraffin-embedded RNA Isolation Kit (Qiagen, Hilden, Germany). RNA quantification was done with a Qubit 2.0 fluorometer (Life Technologies, Carlsbad, CA) and Qubit RNA HS assay kit (Molecular Probes, Eugene, OR). Quality of RNA was assessed by spectrophotometric measurement. JAK2, PD-L1, PD-L2, and PD-1 expression was measured using the Nanostring nCounter PanCancer Immune Profiling Panel (nanoString Technologies) following the manufacturer’s instructions. Data were analyzed by Nanostring nSolver Analysis Software v2.0.
Statistical Analysis
All statistical comparisons and correlations were performed using an unpaired t-test and variation among and between groups was calculated using ANOVA (GraphPad Prism 6). The comparison of array-based comparative genomic hybridization assay and fluorescence in situ hybridization assay P-value was calculated using Pearson’s Chi-square test, and the correlation between the two assays was calculated by Pearson correlation coefficients. P-values≤0.05 was considered significant.
Results
Patients Characteristics
Thirty-four triple negative breast cancer samples were eligible for both array-based comparative genomic hybridization and fluorescence in situ hybridization analysis and the characteristics of the patients are summarized in Table 1. Fifteen samples were classified with copy-number gain at 9p24.1 based on array-based comparative genomic hybridization, and 19 samples were classified as non-gain at 9p24.1. Among these subjects, the majority of patients (n=28/34, 82%) were ≥50 years old, and no significant difference was observed in age distribution between the copy-number gain and non-gain subgroups. The majority of patients had relatively small tumors; 28 subjects (82%) had a primary tumor size of T1-T2. Lymph node status was negative in 40% patients of the gain subgroup and 63% in the non-gain subgroup with no significant difference observed. Most of the patients included in our study were diagnosed with stage I/II breast cancer, with only 5 subjects (15%) with stage III disease. As expected for triple negative breast cancer, the majority of the tumors (n=28, 82%) were grade III. In total, 11 tumors (32%) were derived after neoadjuvant chemotherapy, and were equally distributed in the gain subgroup (n=6, 40%) and in the non-gain subgroup (n=5, 26%). Therefore, these data are likely representative of primary triple negative breast cancer.
Correlation of Array-Based Comparative Genomic Hybridization and Fluorescence in situ Hybridization Classification Of 9p24.1 Copy Number
We have previously reported the copy-number alterations of chromosome 9p24.1 in 34 formalin-fixed paraffin-embedded tumor samples as measured by array-based comparative genomic hybridization.2 Fifteen samples (44%) had 9p24.1 gain and/or amplification and 19 (56%) had non-gain at 9p24.1 (Figure 2a). We then evaluated the novel JAK2 fluorescence in situ hybridization probe, with and without comparison to CEN9, to determine the 9p24.1 copy-number status of these samples. Using the JAK2/CEN9 fluorescence in situ hybridization ratio, the results were 88% concordant with array-based comparative genomic hybridization. Of the 15 samples scored as copy-number gain/amplification by array-based comparative genomic hybridization, 12 samples were concordant by fluorescence in situ hybridization (Figure 2b) and 3 had neutral copy-number (fluorescence in situ hybridization ratio were 1.04, 1.02, and 0.97, separately). In the 19 samples defined as non-gain at 9p24.1 by array-based comparative genomic hybridization, 18 samples were concordant by fluorescence in situ hybridization (Figure 2b). One sample had an average JAK2 signal 1.72 with CEN9 signal of 1.09, scored as gain at 9p24.1 by fluorescence in situ hybridization. No significant difference was found in the distribution of 9p24.1 status in these samples detected by array-based comparative genomic hybridization and fluorescence in situ hybridization (P=0.16) (Table 2). The sensitivity of the fluorescence in situ hybridization test was 80% at 95% specificity when benchmarked to array-based comparative genomic hybridization status (P=0.02).

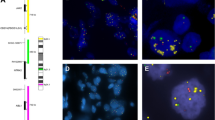
Genomic analysis of the JAK2/9p24.1 locus in triple negative breast cancer. (a) Chromosome 9 copy-number analysis of 3 selected triple negative breast cancer tumors. Locus-specific views of 9p24.1 copy-number variation, red shaded areas denote ADM2 defined copy-number gain/amplification intervals, and green represents deletion. Blue arrows denote the JAK2 locus. Copy-number is scored according to log2 ratio. Top to bottom represent JAK2 amplification, neutral, and deletion (b). Representative of JAK2 status detected by fluorescence in situ hybridization (FISH), JAK2 (red), and chromosome 9 centromere (green). Left: 21+ JAK2 signals and 1–3 CEN9 signals (fluorescence in situ hybridization ratio 8.20, amplification), middle: 2 JAK2 signals and 2 CEN9 signals (fluorescence in situ hybridization ratio 0.99, neutral), and right: 1–2 JAK2 signals and 2–4 CEN9 signals (fluorescence in situ hybridization ratio 0.56, deletion).
In the copy-number gain subgroup (n=15), JAK2 gain/amplification was detected by fluorescence in situ hybridization ranging from 2.39 to 21.0 (mean 4.65). To adjust for aneuploidy, the ratio of JAK2 to CEN9 ranged from 0.78 to 8.2 (mean 1.87). The sample with highest level of amplification (array-based comparative genomic hybridization log2ratio=3.6) as measured by array-based comparative genomic hybridization also scored highest by fluorescence in situ hybridization (fluorescence in situ hybridization ratio=8.2). In the non-amplification subgroup (n=19), average JAK2 signals were detected by fluorescence in situ hybridization with the range from 0.94 to 2.84 (mean 1.67), with the ratio of JAK2:CEN9 ranging from 0.42 to 1.58 (mean 0.75).
In 12 samples with copy-number gain by fluorescence in situ hybridization, 3 samples had polysomy 9 (CEN9 ≥3.0),32, 33 and 1 of 18 samples had polysomy 9. Polysomy 9 has been reported in pancreatic ductal adenocarcinoma and ovarian cancer but the effect on the 9p24.1 transcription and translation is not clear.33 We separately evaluated the average JAK2 signals by fluorescence in situ hybridization and the fluorescence in situ hybridization ratio (average JAK2 signal/CEN9). We independently calculated the correlation of copy-number status as determined by array-based comparative genomic hybridization with the average JAK2 copy-number by fluorescence in situ hybridization, and the JAK2/CEN9 ratio by fluorescence in situ hybridization using the Spearman correlation. We observed a strong correlation of copy-number status by array-based comparative genomic hybridization with both JAK2 and the JAK2/CEN9 ratio (r=0.85, 0.83, P<0.0001; Figure 3).

Comparison of JAK2/9p24.1 copy-number status detected by array-based comparative genomic hybridization and fluorescence in situ hybridization cases are plotted based on the array-based comparative genomic hybridization (aCGH) and fluorescence in situ hybridization (FISH) ratios. (a) The x axis represents array-based comparative genomic hybridization ratio, and the y axis represents fluorescence in situ hybridization ratio, as the ratio of mean JAK2 score/mean CEN9 score. (b) Cases are plotted based on the array-based comparative genomic hybridization and average JAK2 signals detected by fluorescence in situ hybridization. The x axis represents array-based comparative genomic hybridization ratio, the y axis represents average JAK2/9p24.1 signals by fluorescence in situ hybridization. The correlation between array-based comparative genomic hybridization and fluorescence in situ hybridization was calculated by Pearson correlation coefficients.
JAK2 mRNA Expression Correlates with 9p24.1 Copy-number Status
We have previously reported a strong correlation between JAK2 mRNA levels and 9p24.1 amplification using qRT-PCR on a limited set of triple negative breast cancer samples.2 Here, we evaluated the RNA expression of JAK2, PD-L1, and PD-L2 in 17 samples, using Nanostring immune profiling.20 Almost all tumors were treatment-naïve; six samples were collected after neoadjuvant chemotherapy in the copy-number gain (n=4) and non-gain (n=2) subgroups. We observed a correlation between copy-number gain of 9p24.1 and significant elevation of JAK2 mRNA levels (Figure 4a). The level of JAK2 mRNA was 633±229 (mean±s.d.) in the array-based comparative genomic hybridization-defined gain subgroup, higher than 382±160 (mean±s.d.) in the non-gain subgroup (P=0.02) (Table 3). This correlation between gain and non-gain subgroups was also observed using the fluorescence in situ hybridization-defined copy-number status (P=0.05) (Figure 4b), where the mean level of JAK2 mRNA was 627±251 (mean±s.d.) in the fluorescence in situ hybridization -defined gain subgroup and 408±174 (mean±s.d.) in the non-gain subgroup (Table 3). The range of the expression of JAK2 was the same regardless of array-based comparative genomic hybridization or fluorescence in situ hybridization analysis (range 456–1095 in copy-number gain group, and 183–755 in non-gain group) (Table 3). Here, we confirmed that RNA expression of JAK2 is elevated in samples with 9p24.1 copy-number gain.

Association of JAK2/9p24.1 copy-number status with RNA expression of JAK2, PD-L1 and PD-L2 A total of 17 fresh frozen tumor samples were available for measuring RNA expression by Nanostring immunoprofiling. The expression level of JAK2 (a), PD-L1 (b) and PD-L2 (c) were compared between the gain and non-gain groups (grouped by array-based comparative genomic hybridization (aCGH) (left) or fluorescence in situ hybridization (FISH) (right) ratio). P-value was calculated by unpaired Student t-test.
Limited Association of PD-L1 and PD-L2 Expression with 9p24.1 Copy-number Status
In our prior study, 9p24.1 amplification was more variably associated with PD-L1 RNA expression and was not associated with PD-L2 expression.2 Here, we confirmed PD-L1 expression weakly associated with array-based comparative genomic hybridization 9p24.1 status, and with significant variation in expression levels (Figure 4b). In the array-based comparative genomic hybridization-defined gain subgroup, the level of PD-L1 expression was 46±45 (mean±s.d.), compared to 21±9.5 (mean±s.d.) in the non-gain group (P=0.11; Table 3). A weaker trend was observed in the fluorescence in situ hybridization-defined gain and non-gain subgroups (mean±s.d. 43±49 vs 25±15, P=0.25). Compared to the non-gain subgroup, the range of PD-L1 expression in cases with 9p24.1 copy-number gain was highly variable, ranging from 12 to 141 in compared with a more limited range of 9 to 41 within the non-gain subgroup. PD-L1 immunohistochemistry was performed in 33 samples, of which 48% (n=16) were PD-L1 positive. No significant association was observed between PD-L1 immunohistochemistry expression and the copy-number gain status defined by array-based comparative genomic hybridization assay (P=0.3), or fluorescence in situ hybridization assay (P=0.5). Our data support that there are multiple mechanisms beyond copy-number that drive PD-L1 expression and are in concordance with the variability of PD-L1 positivity reported across different cancer types and studies.34
PD-L2 is a second ligand of PD-1, and PD-L2 is tightly linked to JAK2 and PD-L1 within the 9p24.1 chromosomal region. A weak trend of association between the expressing level of PD-L2 and 9p24.1 status was observed (P=0.14) (Figure 4c). In the copy-number gain subgroup as defined by array-based comparative genomic hybridization, the expression of PD-L2 ranged from 23 to 300 (mean±s.d. 145±91), compared to 96±32 (mean±s.d.) in the non-gain subgroup. By fluorescence in situ hybridization analysis, PD-L2 expression was not associated with copy-number status (mean 136±96 s.d. vs mean 106±44, P=0.39) (Table 4). We did not observe an association between the 9p24.1 status and PD-1 expression in our study (data not shown).
Discussion
Triple negative breast cancer remains a clinical challenge despite advances in treatment, and there remains an urgent need for novel therapies.10, 35, 36 Advances in genomics have led to the recognition of functional diversity within the triple negative breast cancer subset of tumors.37, 38 Specific genetic alterations, such copy-number amplification, may serve as predictive biomarkers for targeted treatments.39 High-resolution CGH arrays are a robust, highly quantitative technology to detect copy-number alterations, providing whole chromosome analysis for as small as 70-kb intervals,39 but array-based comparative genomic hybridization technology is not yet feasible for high-throughput copy-number analysis of clinical samples. Due to the strong association of 9p24.1 copy-number status with both response to targeted JAK2 inhibition in vitro and in vivo,2, 4 and the observation in lymphoma that 9p24.1 copy-number may be associated with response to PD-1/PD-L1 checkpoint blockade,40 we sought to develop a robust, clinically-compatible fluorescence in situ hybridization assay for assessing 9p24.1 copy-number status in clinical samples, benchmarked to array CGH status. We focused this study on triple negative breast cancer, where we have observed up to 25% of untreated tumors have evidence of 9p24.1 copy-number gain, which is associated with poor clinical outcome.2 We developed a novel JAK2/9p24.1 fluorescence in situ hybridization probe to quantitate copy-number alterations across primary triple negative breast cancer tumors and compared the results to array-based comparative genomic hybridization using purified aneuploid tumor nuclei.41 We focused our fluorescence in situ hybridization assay development on the JAK2 locus, as we have not yet observed segregation of this locus with the tightly linked PD-L1 locus (n>300 tumors).
As expected, the majority (n=30) of the 34 selected tumors showed concordant copy-number status between array-based comparative genomic hybridization and fluorescence in situ hybridization. We eliminated four samples from analysis either due to low tumor content (n=3) or due to a mixed population of tumor cells with distinct array-based comparative genomic hybridization profiles (n=1, Figure 1). Of the 34 tumors, there were three samples that had borderline-low level copy-number gain by array-based comparative genomic hybridization (ratios 0.3, 0.48 and 0.58, cutoff=0.3) that were measured as copy-number neutral by fluorescence in situ hybridization. In comparison, only one sample that was pre-defined as neutral by array-based comparative genomic hybridization had low level gain by fluorescence in situ hybridization (ratio=1.58), due to a deletion across CEN9 (JAK2=1.72, CEN9=1.09), which led to a higher JAK2/9p24.1 fluorescence in situ hybridization ratio. These data are consistent with the observation that fluorescence in situ hybridization analysis may have a lower overall sensitivity of detection of copy-number than array-based comparative genomic hybridization, but the latter provides both whole chromosome and genome-wide analysis.23 However, high cost and the technical requirements limit the clinical application of array-based comparative genomic hybridization.23, 41 Of the 34 samples included in this analysis, JAK2/9p24.1 fluorescence in situ hybridization analysis identified 88.2% of the JAK2/9p24.1 copy-number status as measured by array-based comparative genomic hybridization on purified tumor nuclei.
One major issue of clinical fluorescence in situ hybridization analysis is the presence of aneuploidy in tumor samples. The importance of using centromeric ratios to assess the copy number of specific genes has been previously recognized,42 which adjusts for the natural increase of copy number during replication and corrects for chromosomal aneuploidy.43 For the HER-2/neu locus, the frequency of polysomy of chromosome 17 has been reported from 13 to 46% in breast cancer, and was not associated with HER-2/neu overexpression on immunohistochemistry or mRNA level.32, 44, 45 In our study, we found 4 out of 34 (12%) samples had polysomy of chromosome 9 in triple negative breast cancer, but the association between this polysomy and the significance to the regulation of JAK2/PD-L1 expression in triple negative breast cancer remains unclear. We detected only one sample with 9p24.1 copy-number gain that included CEN9, resulting in a false negative fluorescence in situ hybridization ratio. In a recent study using a fluorescence in situ hybridization probe mapping to 9p24.1 included PD-L1 to detect copy-number alteration in classical Hodgkin lymphoma, 56% of tumors had 9p24.1 copy gain and 36% had 9p24.1 copy amplification.5 A significant correlation between the 9p24.1 amplification and JAK2 overexpression both at the mRNA and protein levels has been observed in both breast cancer and lymphoma.2, 32, 46 As such, we designed our fluorescence in situ hybridization probe mapping to 9p24.1 to include JAK2, and given the short distance (322 kb), this probe will likely serve as a surrogate for the PD-L1 locus as well.
We have analyzed copy-number alterations at the 9p24.1 locus across over 300 tumors from melanoma, pancreatic cancer, glioblastoma, colorectal cancer, and breast cancer.2 The shortest region of overlap (SRO) maps to a 777 kb region that encodes PD-L1, PD-L2, JAK2 and four insulin-like proteins (INSL6, INSL4, RLN2, and RLN1) which have been implicated in carcinogenesis.2, 47, 48 Inhibitors of JAK2 and PD-L1 are in clinical use, but predictive biomarkers for tumor response are needed. In particular, the strong correlation between 9p24.1 copy-number gain/amplification and JAK2 expression makes chromosome 9p24.1 copy-number status a plausible predictive biomarker candidate for JAK2/STAT3 pathway inhibition.1, 2, 4 The relaxin proteins (RLN1 and RLN2) are secreted and interact with the GCPR relaxin receptors RXFPs (LGR7/8). The relaxin receptor antagonist, AT-001 inhibits human prostate cancer xenografts and is synergistic with docetaxel.49, 50 Targeted relaxin receptor inhibitors are in preclinical development for prostate, breast, and ovarian cancer. Highly homologous to the relaxins, insulin-like peptides (INSL proteins 1–7) are members of the insulin family and are secreted proteins that are expressed mainly in testis, placenta, and uterus. INSL4 (EPIL) functions in the placenta to enhance tissue invasiveness and cell migration, and has been shown to be overexpressed in a subset of breast cancers, and is associated in cell invasion in vitro.51 The JAK2/9p24.1 probe set may be useful for analysis copy number of these other tightly-linked genes.
We observed that RNA expression of JAK2 is correlated with JAK2/9p24.1 copy-number status. Of the 17 samples in our study that were assessed for RNA expression, 4 samples were collected after neoadjuvant chemotherapy. Both 9p24.1 amplification is enriched in breast cancer after neadjuvant therapy4 and paclitaxel has been shown to upregulate PD-L1 expression in breast cancer.52 We did not observe a signficant difference in copy-number distribution with or without prior neoadjuvant chemotherapy, but the sample size is limited. However, we did observe that the highest JAK2 and PD-L1 mRNA levels were detected in the tissue samples obtained after neoadjuvant chemotherapy.
Multiple studies have addressed the limitations of the PD-L1 immunohistochemistry assay as a predictive biomarker for response to checkpoint blockade.34, 53 The expression of PD-L1 is dynamically regulated by multiple mechanisms and is detected on both tumor and immune cells.33 Different antibodies to PD-L1, and different thresholds have been applied to define PD-L1 expression by immunohistochemistry.52 In our study, we did not observe a significant association between the PD-L1 immunohistochemistry status and copy-number gain, likely reflecting a complex regulatory network of PD-L1 expression.
Our study benchmarks a novel fluorescence in situ hybridization probe localizing to the JAK2 locus on chromosome 9p24.1 to array-based comparative genomic hybridization status using a highly-purified tumor population, controlling for aneuploidy at chromosome 9 using a centromeric probe. This may serve as a clinically relevant biomarker for targeting JAK2 and PD-L1 inhibitors, as well as the tightly-linked insulin-like peptide genes that are present on this locus. Further studies are warranted to assess optimal cutoff levels of JAK2 fluorescence in situ hybridization, the clinical association of therapeutic response with copy-number status at 9p24.1, as well as to further elucidate the tumor biology underlying the more variable expression of PD-L1.
References
Hao Y, Chapuy B, Monti S et al. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res 2014;20:2674–2683.
Barrett MT, Anderson KS, Lenkiewicz E et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015;6:26483–26493.
Wu J, Liu S, Liu G et al. Identification and functional analysis of 9p24 amplified genes in human breast cancer. Oncogene 2012;31:333–341.
Balko JM, Schwarz LJ, Luo N et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci Transl Med 2016;8:334ra353.
Roemer MG, Advani RH, Ligon AH et al. PD-L1 and PD-L2 genetic alterations define classical Hodgkin lymphoma and predict outcome. J Clin Oncol 2016;34:2690–2697.
Kim MS, Lee WS, Jeong J et al. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 2015;6:40158–40171.
Momtaz P, Postow MA . Immunologic checkpoints in cancer therapy: focus on the programmed death-1 (PD-1) receptor pathway. Pharmgenomics Pers Med 2014;7:357–365.
Sun WY, Lee YK, Koo JS . Expression of PD-L1 in triple-negative breast cancer based on different immunohistochemical antibodies. J Transl Med 2016;14:173.
Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–2454.
Lipson EJ, Forde PM, Hammers HJ et al. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin Oncol 2015;42:587–600.
Tavallai M, Booth L, Roberts JL et al. Ruxolitinib synergizes with DMF to kill via BIM+BAD-induced mitochondrial dysfunction and via reduced SOD2/TRX expression and ROS. Oncotarget 2016;7:17290–17300.
Xin H, Herrmann A, Reckamp K et al. Antiangiogenic and antimetastatic activity of JAK inhibitor AZD1480. Cancer Res 2011;71:6601–6610.
Harrison CN, Vannucchi AM, Kiladjian JJ et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016;30:1701–1707.
Quintas-Cardama A, Verstovsek S . Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res 2013;19:1933–1940.
Patel KP, Newberry KJ, Luthra R et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015;126:790–797.
Cimino-Mathews A, Thompson E, Taube JM et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum Pathol 2016;47:52–63.
Lastwika KJ, Wilson W 3rd, Li QK et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res 2016;76:227–238.
Mittendorf EA, Philips AV, Meric-Bernstam F et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2014;2:361–370.
Taube JM, Klein A, Brahmer JR et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 2014;20:5064–5074.
Lee HJ, Lee JJ, Song IH et al. Prognostic and predictive value of NanoString-based immune-related gene signatures in a neoadjuvant setting of triple-negative breast cancer: relationship to tumor-infiltrating lymphocytes. Breast Cancer Res Treat 2015;151:619–627.
Pinkel D, Segraves R, Sudar D et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998;20:207–211.
Bejjani BA, Shaffer LG . Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn 2006;8:528–533.
Yeh IT, Martin MA, Robetorye RS et al. Clinical validation of an array CGH test for HER2 status in breast cancer reveals that polysomy 17 is a rare event. Mod Pathol 2009;22:1169–1175.
Barrans SL, Evans PA, O’Connor SJ et al. The detection of t(14;18) in archival lymph nodes: development of a fluorescence in situ hybridization (FISH)-based method and evaluation by comparison with polymerase chain reaction. J Mol Diagn 2003;5:168–175.
Bartlett JM, Going JJ, Mallon EA et al. Evaluating HER2 amplification and overexpression in breast cancer. J Pathol 2001;195:422–428.
Lipson D, Aumann Y, Ben-Dor A et al. Efficient calculation of interval scores for DNA copy number data analysis. J Comput Biol 2006;13:215–228.
Hodge JC, Bedroske PP, Pearce KE et al. Molecular cytogenetic analysis of JAZF1, PHF1, and YWHAE in endometrial stromal tumors: discovery of genetic complexity by fluorescence in situ hybridization. J Mol Diagn 2016;18:516–526.
Romano RC, Shon W, Sukov WR . Malignant melanoma of the nail apparatus: a fluorescence in situ hybridization analysis of 7 cases. Int J Surg Pathol 2016;24:512–518.
Brockman SR, Paternoster SF, Ketterling RP et al. New highly sensitive fluorescence in situ hybridization method to detect PML/RARA fusion in acute promyelocytic leukemia. Cancer Genet Cytogenet 2003;145:144–151.
Choschzick M, Lassen P, Lebeau A et al. Amplification of 8q21 in breast cancer is independent of MYC and associated with poor patient outcome. Mod Pathol 2010;23:603–610.
Sabbatino F, Villani V, Yearley JH et al. PD-L1 and HLA Class I antigen expression and clinical course of the disease in intrahepatic cholangiocarcinoma. Clin Cancer Res 2016;22:470–478.
Vanden Bempt I, Van Loo P, Drijkoningen M et al. Polysomy 17 in breast cancer: clinicopathologic significance and impact on HER-2 testing. J Clin Oncol 2008;26:4869–4874.
Luo Y, Tian L, Feng Y et al. The predictive role of p16 deletion, p53 deletion, and polysomy 9 and 17 in pancreatic ductal adenocarcinoma. Pathol Oncol Res 2013;19:35–40.
Patel SP, Kurzrock R . PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–856.
Cleator S, Heller W, Coombes RC . Triple-negative breast cancer: therapeutic options. Lancet Oncol 2007;8:235–244.
Ribas A, Tumeh PC . The future of cancer therapy: selecting patients likely to respond to PD1/L1 blockade. Clin Cancer Res 2014;20:4982–4984.
Shaver TM, Lehmann BD, Beeler JS et al. Diverse, biologically relevant, and targetable gene rearrangements in triple-negative breast cancer and other malignancies. Cancer Res 2016;76:4850–4860.
Schneider BP, Winer EP, Foulkes WD et al. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 2008;14:8010–8018.
Andre F, Job B, Dessen P et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 2009;15:441–451.
Ansell SM, Lesokhin AM, Borrello I et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2015;372:311–319.
Ruiz C, Lenkiewicz E, Evers L et al. Advancing a clinically relevant perspective of the clonal nature of cancer. Proc Natl Acad Sci U S A 2011;108:12054–12059.
Dal Lago L, Durbecq V, Desmedt C et al. Correction for chromosome-17 is critical for the determination of true Her-2/neu gene amplification status in breast cancer. Mol Cancer Ther 2006;5:2572–2579.
Tse CH, Hwang HC, Goldstein LC et al. Determining true HER2 gene status in breast cancers with polysomy by using alternative chromosome 17 reference genes: implications for anti-HER2 targeted therapy. J Clin Oncol 2011;29:4168–4174.
Rosenberg CL . Polysomy 17 and HER-2 amplification: true, true, and unrelated. J Clin Oncol 2008;26:4856–4858.
Press MF, Bernstein L, Thomas PA et al. HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J Clin Oncol 1997;15:2894–2904.
Soliman H, Khalil F, Antonia S . PD-L1 expression is increased in a subset of basal type breast cancer cells. PLoS One 2014;9:e88557.
Van Der Westhuizen ET, Summers RJ, Halls ML et al. Relaxin receptors—new drug targets for multiple disease states. Curr Drug Targets 2007;8:91–104.
Sferruzzi-Perri AN, Owens JA, Standen P et al. Early pregnancy maternal endocrine insulin-like growth factor I programs the placenta for increased functional capacity throughout gestation. Endocrinology 2007;148:4362–4370.
Neschadim A, Pritzker LB, Pritzker KP et al. Relaxin receptor antagonist AT-001 synergizes with docetaxel in androgen-independent prostate xenografts. Endocr Relat Cancer 2014;21:459–471.
Neschadim A, Summerlee AJ, Silvertown JD . Targeting the relaxin hormonal pathway in prostate cancer. Int J Cancer 2014;137:2287–2295.
Brandt B, Roetger A, Bidart JM et al. Early placenta insulin-like growth factor (pro-EPIL) is overexpressed and secreted by c-erbB-2-positive cells with high invasion potential. Cancer Res 2002;62:1020–1024.
Zhang P, Su DM, Liang M et al. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol Immunol 2008;45:1470–1476.
Gibney GT, Weiner LM, Atkins MB . Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol 2016;17:e542–e551.
Acknowledgements
We thank the Mayo Cytogenetics Core, Dr Patricia T. Greipp, Sara Kloft-Nelson, Darlene Knutson and Ryan Knudson.We also thank Niro Ramachandran at Nanostring Technologies for assistance with the sample analysis. This work was supported by the Breast Cancer Research Foundation (KSA) and the CARE Foundation (BAP, MTB, and KSA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
KSA has served as a consultant and has received stock options from Provista Dx, and is a founder of FlexBioTech. LA and JHY are full time employees at Merck Research Laboratories. The remaining authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Chen, M., Andreozzi, M., Pockaj, B. et al. Development and validation of a novel clinical fluorescence in situ hybridization assay to detect JAK2 and PD-L1 amplification: a fluorescence in situ hybridization assay for JAK2 and PD-L1 amplification. Mod Pathol 30, 1516–1526 (2017). https://doi.org/10.1038/modpathol.2017.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.86
This article is cited by
-
Mapping cancer biology in space: applications and perspectives on spatial omics for oncology
Molecular Cancer (2024)
-
PD-L1 amplification is associated with an immune cell rich phenotype in squamous cell cancer of the lung
Cancer Immunology, Immunotherapy (2021)
-
Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours
Nature Biomedical Engineering (2019)
-
JAK2/PD-L1/PD-L2 (9p24.1) amplifications in renal cell carcinomas with sarcomatoid transformation: implications for clinical management
Modern Pathology (2019)
-
A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma
Nature Medicine (2019)
