
Abstract
Esophageal epidermoid metaplasia is a rare condition that involves the proximal-to-middle third of the esophagus. It is sharply demarcated and defined histologically by epithelial hyperplasia, a prominent granular cell layer, and superficial hyperorthokeratosis. In addition, preliminary studies have suggested an association between esophageal epidermoid metaplasia and esophageal squamous neoplasia (squamous dysplasia and esophageal squamous cell carcinoma). To further characterize esophageal epidermoid metaplasia and better define its relationship to squamous neoplasia of the esophagus, we performed targeted next-generation sequencing on uninvolved esophageal squamous mucosa and matching esophageal epidermoid metaplasia specimens from 18 patients. Further, we evaluated both synchronous and metachronous high-grade squamous dysplasia/esophageal squamous cell carcinoma by next-generation sequencing from 5 of the 18 (28%) patients, and compared these findings to corresponding esophageal epidermoid metaplasia specimens. Targeted next-generation sequencing revealed 12 of 18 (67%) esophageal epidermoid metaplasia specimens’ harbored alterations in genes often associated with esophageal squamous cell carcinoma. The most frequently mutated genes consisted of TP53 (n=10), PIK3CA (n=2), EGFR (n=2), MYCN (n=1), HRAS (n=1), and the TERT promoter (n=1). Sequencing of synchronous and metachronous high-grade squamous dysplasia/esophageal squamous cell carcinoma identified shared genetic alterations with corresponding esophageal epidermoid metaplasia specimens that suggests a clonal relationship between these entities. In addition, the presence of a TP53 mutation in esophageal epidermoid metaplasia specimens correlated with concurrent or progression to high-grade squamous dysplasia/esophageal squamous cell carcinoma. No genetic alterations were detected in uninvolved esophageal squamous mucosa. On the basis of these findings, we conclude esophageal epidermoid metaplasia is a precursor to in situ and invasive esophageal squamous neoplasia. Further, the detection of TP53 mutations in esophageal epidermoid metaplasia specimens may serve as an early detection biomarker for high-grade squamous dysplasia/esophageal squamous cell carcinoma.
Similar content being viewed by others


Main
Esophageal cancer is the 8th most common cancer and 6th leading cause of cancer-related deaths in the world.1, 2 The overall prognosis of esophageal cancer is poor with a 5-year survival that ranges from 15 to 25%.3, 4 However, improved patient outcomes have been reported when esophageal cancer is diagnosed in its early stages. Cancers of the esophagus are primarily categorized into two subtypes: esophageal squamous cell carcinoma and esophageal adenocarcinoma. Although the incidence of esophageal adenocarcinoma has increased rapidly, esophageal squamous cell carcinoma remains the predominant form of esophageal cancer worldwide and accounts for 80% of all cases.2, 5 Risk factors for esophageal squamous cell carcinoma include increased alcohol consumption, tobacco use, nutritional deficiencies, poor oral hygiene, and thermal damage.5 Despite the broad etiologic origins of esophageal squamous cell carcinoma, esophageal squamous cell carcinoma is believed to arise from common premalignant precursor lesions. For example, squamous dysplasia of the esophagus is often seen in resection specimens of esophageal squamous cell carcinoma and in patients at a high-risk for developing esophageal squamous cell carcinoma.6, 7 In addition, follow-up studies of patients with untreated esophageal squamous dysplasia have shown an increased frequency of esophageal squamous cell carcinoma.8 Thus, recognition of precursor lesions to esophageal squamous cell carcinoma and esophageal squamous dysplasia would be of interest in identifying high-risk patients and designing preventive screening protocols.
Recently, we and others have reported the clinicopathologic features of a rare condition involving the proximal-to-middle third of the esophagus termed esophageal epidermoid metaplasia.9, 10, 11, 12, 13, 14, 15 Endoscopically, esophageal epidermoid metaplasia is often described as a sharply demarcated patch or plaque consisting of white-to-tan mucosa. Biopsies of esophageal epidermoid metaplasia demonstrate a thickened basal layer, an acanthotic midzone, a prominent granular cell layer, which is normally absent in the esophageal squamous mucosa, and superficial hyperorthokeratosis. The etiology of esophageal epidermoid metaplasia is unclear, but patients with esophageal epidermoid metaplasia frequently report a long-term increased alcohol consumption and tobacco use, similar to patients with esophageal squamous cell carcinoma.9, 10, 15 Moreover, previous studies have found a high rate of both synchronous and metachronous esophageal squamous dysplasia, and esophageal squamous cell carcinoma in patients with esophageal epidermoid metaplasia.9, 15 These findings suggest an association between esophageal epidermoid metaplasia, and esophageal squamous dysplasia and esophageal squamous cell carcinoma. However, whether esophageal epidermoid metaplasia represents a precursor lesion to esophageal squamous dysplasia and/or esophageal squamous cell carcinoma is unknown and merits further investigation.
The accumulation of genetic alterations has a pivotal role during malignant progression of normal esophageal squamous mucosa to esophageal squamous cell carcinoma. Candidate gene sequencing, allelotyping with polymorphic genetic markers, comparative genomic hybridization, and whole-genome/exome sequencing have identified numerous molecular abnormalities in the pathogenesis of esophageal squamous cell carcinoma.16, 17, 18, 19, 20, 21, 22 These studies implicate a number of well-known tumor suppressor genes and oncogenes, such as TP53, CDKN2A, PIK3CA, RB1, and genes within the receptor tyrosine kinase/mitogen-activated protein kinase pathways. Therefore, to further characterize esophageal epidermoid metaplasia and define its relationship to squamous neoplasia of the esophagus, we performed targeted next-generation sequencing of esophageal epidermoid metaplasia specimens and matching uninvolved esophageal squamous mucosa for genes commonly altered in malignant neoplasms. In addition, we evaluated both synchronous and metachronous esophageal squamous dysplasia and esophageal squamous cell carcinoma from the same patients by next-generation sequencing and compared these findings to corresponding esophageal epidermoid metaplasia specimens and uninvolved esophageal squamous epithelium.
Materials and methods
Study Population
Study approval was obtained from the University of Pittsburgh and Johns Hopkins University Institutional Review Boards. The anatomic pathology archives from the Departments of Pathology at the University of Pittsburgh Medical Center and Johns Hopkins Hospital were queried for the diagnosis of esophageal epidermoid metaplasia or documentation of esophageal biopsies demonstrating hyperorthokeratosis and a prominent granular cell layer.9 Corresponding hematoxylin-and-eosin stained slides for cases identified with the aforementioned criteria were reviewed and, in total, 18 patients with esophageal epidermoid metaplasia were identified. Formalin-fixed paraffin-embedded tissue blocks for all 18 cases of esophageal epidermoid metaplasia and uninvolved esophageal mucosa (n=36) were retrieved for molecular analysis. These cases consisted of 17 pairs of esophageal mucosal biopsies and 1 resection specimen. Corresponding synchronous and metachronous esophageal squamous cell carcinoma and high-grade squamous dysplasia specimens were also identified from the same patients. Formalin-fixed paraffin-embedded tissue blocks were retrieved and also submitted for molecular analysis (n=5). Patient demographic data, endoscopic reports, and follow-up information were also recorded. Clinically significant tobacco smoking and alcohol consumption were defined as >10 pack year history and >2 drinks per day, respectively.
Molecular Testing
A total of 41 specimens were submitted to the Molecular and & Genomic Pathology Laboratory at University of Pittsburgh Medical Center for targeted next-generation sequencing. From formalin-fixed paraffin-embedded tissues, uninvolved esophageal mucosa, esophageal epidermoid metaplasia, and high-grade squamous dysplasia/esophageal squamous cell carcinoma were microdissected from eight 4-μm unstained histologic sections under stereomicroscopic visualization with an Olympus SZ61 microscope (Olympus). Genomic DNA was isolated from the DNeasy Blood and Tissue Kit on the automated QIAcube (Qiagen) instrument according to the manufacturer’s instructions. Extracted DNA was quantitated on the Qubit 2.0 Fluorometer using the dsDNA HS Assay Kit (Thermo Fisher Scientific).
Amplification-based targeted next-generation sequencing was performed using custom primer pools designed with Life Technologies’ design tool. Custom libraries included 396 total primer pairs in 2 primer pools for amplification and sequencing of the following genes: AKT1, ATRX, BRAF, CDK6, CDKN2A, CIC, CTNNB1, DDX3X, EGFR, FUBP1, H3F3A, HRAS, IDH1, IDH2, KLF4, KRAS, MET, MYC, MYCN, NF1, NF2, NRAS, PIK3CA, PTCH1, PTEN, RB1, SETD2, SMO, TERT, and TP53.23 Overall, 10–20 ng of DNA were amplified by PCR using the premixed primer pools and Ion AmpliSeq HiFi Master Mix (Ion AmpliSeq Library Kit 2.0). Amplicons were treated with FuPa reagent to partially digest primer sequences and phosphorylate the amplicons. The amplicons were then ligated to adapters with the addition of barcodes from the Ion Xpress Barcode Adapters 1–96 Kit according to manufacturer’s instructions (Life Technologies). After ligation, the amplicons underwent nick-translation and additional library amplification by PCR to complete the linkage between adapters and amplicons. Library concentration and amplicon size were determined using the Tapestation 2200 (Agilent). Equal molar volumes of libraries from the two primer pools were combined, and multiplexed barcoded libraries were enriched by clonal amplification using emulsion PCR on Ion Sphere particles (Ion PGMTemplate OT2 200 kit or Ion PI OT2 200 kit v3) and loaded on an Ion 318 Chip or Ion P1 Chip (Life Technologies). Massively parallel sequencing was carried out on a Personal Genome Machine Sequencer (Ion Torrent) or Ion Proton according to the manufacturer’s instructions (Life Technologies). Validation of this panel was performed as recommended by the College of American Pathologists to establish analytical accuracy of the assay.24 The analytical sensitivity or limits of detection was determined using serial dilutions of brain tumor DNA diluted in DNA from normal tissue. The limit of detection was 5% mutational allelic frequency at 500 × or 3% mutational allelic frequency at 1000 × coverage for each tested region. The minimum depth of coverage was 500 ×.
Raw signal data were analyzed using Torrent Suite (version 4.0.1) to generate (.bam) files after signal processing, base-calling adapter trimming, and alignment to the reference human genome (hg19). Variants were called with Torrent Suite Variant Caller and were further analyzed using an internally created software suite based on ANNOVAR, which annotates each variant with SIFT20 and Polyphen-221 scores predicting its effect on the protein function, and with existing databases including dbSNP22 and COSMIC.25, 26, 27, 28, 29 For each mutation identified, a mutational allelic frequency was calculated based on the number of reads of the mutant allele vs the wildtype allele and reported as a percentage. Analysis of copy number alterations and establishment of the copy number ratio was performed as previously reported.30
Fluorescence In Situ Hybridization
Dual color fluorescence in situ hybridization was performed for CDKN2A, as previously reported.31 CDKN2A was assessed using a Spectrum-Orange labeled, locus-specific probe (Abbott Molecular) with a Spectrum Green-labeled chromosome 9 centromeric probe. Staining was performed as previously described using 4-μm unstained formalin-fixed paraffin-embedded sections. At least 50 epithelial cells were assessed for each specimen and scored as a ratio of the average CDKN2A copy number per cell to the average chromosome 9 centromeric probe copy number.
Statistical Analysis
Chi-squared analysis or Fisher exact tests were used to compare categorical data, and analysis of variance was used to compare continuous variables. All statistical analyses were performed using the SPSS Statistical software, version 21 (IBM, Armonk, NY, USA) and statistical significance was defined as a P-value of <0.05.
Results
Patient Cohort
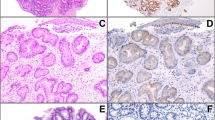
The clinical, endoscopic, and associated pathologic findings of the study cohort are summarized in Table 1. Patients at initial diagnosis ranged in age from 47 to 87 years (mean, 68.2 years; median, 67 years) with a slight male predominance (10 of 18, 56%). The past medical history was significant for tobacco smoking in 13 (72%) patients, increased alcohol intake in 6 (33%), head and neck squamous cell carcinoma in 1 (6%), lichenoid esophagitis in 1 (6%), and esophageal lichen planus in 1 (6%).32 Endoscopically, esophageal epidermoid metaplasia appeared as white-to-tan plaques (Figure 1a). These lesions ranged in size from 1 to 24 cm in greatest dimension (mean, 8.5 cm; median, 8.5 cm) and were located throughout the esophagus, with the majority located in the proximal-to-middle third. Consistent with prior studies, each case of esophageal epidermoid metaplasia was histologically characterized by an undulating and hyperplastic squamous mucosa with flattening of the rete pegs, a prominent granular cell layer and compact hyperorthokeratosis (Figure 1b and c). Other pathologic findings within the patient cohort included an adjacent esophageal squamous cell carcinoma (n=1, Patient 1) and high-grade squamous dysplasia (n=2, Patients 3 and 4) (Figure 1d). For one patient, Barrett’s mucosa without dysplasia was present in the distal esophagus.

Endoscopic images of esophageal epidermoid metaplasia reveal well-demarcated areas of white, cobblestone appearing patches within the proximal-to-middle third of the esophagus (a). Microscopically, esophageal epidermoid metaplasia is characterized by acanthotic squamous mucosa, a prominent granular cell layer, and superficial hyperorthokeratosis (b). In addition, esophageal epidermoid metaplasia is associated with synchronous and metachronous high-grade squamous dysplasia (c) and esophageal squamous cell carcinoma (d) in a subset of cases.
Follow-up information was available for 15 of 18 (83%) patients and ranged from 1 to 40 months (mean, 18 months). A minimally invasive esophagectomy was performed for patient 1. Subsequent pathology of the esophagectomy specimen revealed a 3 cm squamous cell carcinoma of the distal esophagus extending into the submucosa with negative regional lymph nodes (pT1bN0). Patients 3 and 4 underwent endoscopic mucosal resection for high-grade squamous dysplasia and radiofrequency ablation of the surrounding esophageal epidermoid metaplasia. Continued endoscopic follow-up for these three patients at 24, 12, and 12 months, showed no evidence of esophageal epidermoid metaplasia, dysplasia, or esophageal squamous cell carcinoma. Patient 2 developed an esophageal squamous cell carcinoma and patient 5 developed high-grade squamous dysplasia in the mid esophagus at 36 and 40 months, respectively. Among the remaining 10 patients, 8 had persistent esophageal epidermoid metaplasia on follow-up endoscopy, but none of these patients developed esophageal squamous cell carcinoma or esophageal squamous dysplasia. Patient 6 has also undergone extensive cryotherapy to ‘ablate’ his esophageal epidermoid metaplasia, which has persisted on follow-up.
Paired Analysis of Esophageal Epidermoid Metaplasia and Distant Esophageal Squamous Mucosa
Targeted next-generation sequencing was performed on 18 matched samples of esophageal epidermoid metaplasia and uninvolved esophageal squamous mucosa (Table 2). None of the specimens contained esophageal squamous dysplasia or squamous cell carcinoma. Genetic alterations were detected in 12 (67%) esophageal epidermoid metaplasia cases, but were distinctly absent in paired distant esophageal squamous mucosa from the same patients. The most frequently mutated gene was TP53 (10 of 12, 83%), followed by PIK3CA (n=2), EGFR (n=2), MYCN (n=1), HRAS (n=1), and the TERT promoter (n=1). Missense and nonsense mutations were identified in TP53 that spanned codons 110–282. Further, 3 of 10 TP53-mutated cases harbored 2 different missense mutations in TP53. Activating mutations were detected in PIK3CA (c.1633G>A and c.2176G>A), EGFR (c.865G>A), MYCN (c.793G>A), HRAS (c.34G>A), and the TERT promoter (−124C>T). The mutational allelic frequencies for each gene varied broadly. TP53, PIK3CA, and MYCN mutational allelic frequencies were 3–6%, 3–8%, and 3%, respectively; whereas EGFR, HRAS, and the TERT promoter mutational allelic frequencies were 46–48%, 33%, and 48%, respectively. There were no statistically significant differences between esophageal epidermoid metaplasia with and without genetic alterations with respect to mean patient age (P=0.97), gender (P=0.64), tobacco smoking (P=0.62), alcohol use (P=1.00), and mean esophageal epidermoid metaplasia size (P=0.12).
Paired Analysis of Esophageal Epidermoid Metaplasia and Synchronous and Metachronous High-Grade Squamous Dysplasia and Esophageal Squamous Cell Carcinoma
In addition to paired esophageal epidermoid metaplasia and distant esophageal squamous mucosa specimens, samples from concurrent esophageal squamous cell carcinoma (patient 1) and high-grade squamous dysplasia (patients 3 and 4), and follow-up esophageal squamous cell carcinoma (patient 2) and high-grade squamous dysplasia (patient 5) were assessed for genetic alterations. TP53 was the most frequently mutated gene and missense or nonsense mutations were present in all 5 cases of high-grade squamous dysplasia and esophageal squamous cell carcinoma. Two of 5 cases harbored 2 different missense TP53 mutations, and both corresponded to high-grade squamous dysplasia. Comparative analysis of matched esophageal epidermoid metaplasia specimens revealed at least 1 TP53 mutation was common between both esophageal epidermoid metaplasia, and associated high-grade squamous dysplasia and esophageal squamous cell carcinoma. The mutational allelic frequencies of these shared TP53 mutants in high-grade squamous dysplasia and esophageal squamous cell carcinoma was 33–39% and 10–14%, respectively. Activating mutations in PIK3CA (c.2176G>A) and MYCN (c.793G>A) were also detected in a single case of esophageal squamous cell carcinoma (patient 1) with mutational allelic frequencies of 36 and 21%, respectively. In contrast to esophageal epidermoid metaplasia, homozygous deletions in CDKN2A were identified in both esophageal squamous cell carcinomas and confirmed by fluorescence in situ hybridization. Further, a missense mutation at the acceptor splice site of the CDKN2A intron 1 (c.151-1G>C) was identified in a single case of high-grade squamous dysplasia (patient 3) with a mutational allelic frequency of 8%.
Identification of synchronous and metachronous high-grade squamous dysplasia, and squamous cell carcinoma in esophageal epidermoid metaplasia patients did not correlate with mean patient age (P=0.46), gender (P=0.61), tobacco smoking (P=1.00), alcohol use (P=1.00), mean esophageal epidermoid metaplasia size (P=0.06), or the presence of genetic alterations in matched esophageal epidermoid metaplasia specimens (P=0.11). However, the detection of TP53 mutations in esophageal epidermoid metaplasia was associated with concurrent and progression to high-grade squamous dysplasia and esophageal squamous cell carcinoma (P=0.04).
Discussion
Among other biological mechanisms, it is widely accepted that somatic genetic alterations lead to tumor development in esophageal squamous cell carcinoma. It is postulated that most genetic alterations within a tumor are undamaging byproducts of tumorigenesis (passengers), whereas only a few are responsible for driving tumor initiation and progression (drivers). In esophageal squamous cell carcinoma, a number of genetic alterations have been proposed as drivers, such as inactivation of tumor suppressor genes, TP53 and CDKN2A, or activating mutations in genes involving the receptor tyrosine kinase/mitogen-activated protein kinase pathway.19, 20, 22 Herein, we found 67% of esophageal epidermoid metaplasia specimens harbored alterations in genes often described as drivers of esophageal squamous cell carcinoma or squamous cell carcinomas arising at other sites. Mutations in TP53 were the most frequent genetic alteration in esophageal epidermoid metaplasia and present in 56% of cases. In a subset of esophageal epidermoid metaplasia specimens, more than one TP53 mutation was identified. In addition, paired molecular analysis of esophageal epidermoid metaplasia and synchronous and metachronous high-grade squamous dysplasia/esophageal squamous cell carcinoma revealed at least one TP53 mutation that was identical in both specimens. In comparison, no genetic alterations were detected in the uninvolved esophageal squamous mucosa.
The tumor suppressor gene TP53 encodes for a nuclear phosphoprotein that is involved in DNA repair, cell cycle control, apoptosis, and a multitude of other functions. In esophageal squamous cell carcinoma, inactivation of TP53 is a common genetic event and is reported in 59–93% of tumors.5 Analogous to esophageal epidermoid metaplasia, patients with TP53-mutant esophageal squamous cell carcinoma often report a long history of alcohol consumption and tobacco use.33, 34 Sequencing studies of esophageal squamous cell carcinoma and adjacent esophageal squamous dysplasia have revealed identical TP53 mutations and suggest TP53 alterations occur early in the pathogenesis of esophageal squamous cell carcinoma.35, 36 Moreover, multiple and different TP53 mutations have been found among multifocal squamous dysplasia of the esophagus.37 These findings have led to the concept of field cancerization or field effect for esophageal squamous cell carcinoma.38, 39
The field effect theory postulates that one or more mucosal areas or fields of the esophagus become genomically unstable and predisposed to neoplasia due to prolonged exposure to carcinogens. Additional genetic changes are needed to transform a field into carcinoma. Therefore, the field and carcinoma share genetic alterations and should be considered as having a common clonal origin. The clinical implication of a field is that it may be a source of local recurrences and/or secondary primary carcinomas. The presence of esophageal epidermoid metaplasia may represent a pathologic manifestation of a field in the esophagus. In fact, multiple mutations with a low mutational allelic frequency were identified in esophageal epidermoid metaplasia specimens. Further, an identical TP53 mutation with a higher mutational allelic frequency than esophageal epidermoid metaplasia alone was found in corresponding high-grade squamous dysplasia/esophageal squamous cell carcinoma and, thus, indicating a clonal relationship between esophageal epidermoid metaplasia and the development of esophageal squamous neoplasia.
Despite the frequent presence of TP53 mutations, it is doubtful that TP53 inactivation alone leads to the formation of esophageal epidermoid metaplasia. Considering that mutant TP53 was detected at a low mutational allelic frequency of <6% and not universally present, epithelial cells containing TP53 mutations likely represent subclones within esophageal epidermoid metaplasia. In contrast, esophageal epidermoid metaplasia cases harboring EGFR and HRAS mutations were identified with a mutational allelic frequency of 46–48% and 33%, respectively. As activating mutations in EGFR and HRAS are typically heterozygous, the high mutational allelic frequency would suggest these genetic alterations are present in the majority of the epithelial cells and may drive the pathogenesis of esophageal epidermoid metaplasia. Both EGFR and HRAS are members of the receptor tyrosine kinase/mitogen-activated protein kinase pathway, and similar to TP53 have an early role in tumorigenesis.
The receptor tyrosine kinase/mitogen-activated protein kinase pathway comprises several subpathways that are associated with neoplastic transformation and cancer progression.40 Dysregulation of the receptor tyrosine kinase/mitogen-activated protein kinase pathway occurs in >75% of esophageal squamous cell carcinomas through gene amplification or receptor tyrosine kinase overexpression, chromosomal translocations to produce constitutively active receptor tyrosine kinases, gain of function mutations or deletions that promote ligand-independent receptor tyrosine kinase activity.19, 20, 22 By immunohistochemistry, 50–70% of esophageal squamous cell carcinomas express EGFR protein, whereas 15–28% of tumors exhibit EGFR gene amplification when examined by fluorescence in situ hybridization.41, 42 Further, high mRNA and protein expression of EGFR family members have been reported in so-called oral leukoplakia, an analogous clinicopathologic condition to esophageal epidermoid metaplasia, but in the oral cavity.43, 44 However, activating mutations in EGFR and HRAS comprise a minority of esophageal epidermoid metaplasia cases and, to date, have not been reported in esophageal squamous cell carcinoma. It should be reiterated that aberrant receptor tyrosine kinase/mitogen-activated protein kinase signal transduction may occur through multiple genetic and epigenetic mechanisms. Our study did not comprehensively analyze other members of the receptor tyrosine kinase/mitogen-activated protein kinase pathway that may also be involved in the pathogenesis of esophageal epidermoid metaplasia. It is also important to note that both esophageal epidermoid metaplasia and esophageal squamous cell carcinoma arising in a background of esophageal epidermoid metaplasia are rare (or frequently overlooked) conditions, and previous studies examining the genetics of esophageal squamous cell carcinoma may not have included these cases.
Regardless of its genetic origins, the high rate of esophageal squamous neoplasia in patients with esophageal epidermoid metaplasia, the frequent finding that esophageal epidermoid metaplasia harbors alterations in genes commonly associated with esophageal squamous cell carcinoma, and the clonal relationship between esophageal epidermoid metaplasia and high-grade squamous dysplasia/esophageal squamous cell carcinoma would support esophageal epidermoid metaplasia as a precursor lesion to esophageal squamous neoplasia. Although limited data are available, we and others have recommended endoscopic surveillance for patients with esophageal epidermoid metaplasia.9, 15 Two patients within this study developed high-grade squamous dysplasia/esophageal squamous cell carcinoma on follow-up surveillance. In addition, for a subset of patients, short-interval surveillance and possibly therapeutic intervention, such as endoscopic mucosal resection or radiofrequency ablation, may be warranted. The identification of TP53 mutations in esophageal epidermoid metaplasia specimens was statistically associated with synchronous and metachronous esophageal squamous neoplasia. Thus, mutations in TP53 may serve as a biomarker for early detection of squamous neoplasia in esophageal epidermoid metaplasia patients that can be feasibly tested on routine clinical samples by next-generation sequencing; however, a larger prospective analysis is certainly required.
The present study is not without limitations. It is retrospective by design and, although a large number of specimens were sequenced, the study cohort was relatively small. As discussed previously, targeted next-generation sequencing was limited to known genes frequently associated with malignant neoplasms, and a complete assessment of the entire genome of esophageal epidermoid metaplasia and corresponding esophageal squamous neoplasia was not performed. However, whole-genome sequencing and whole-exome sequencing can be challenging with mucosal biopsies due to limited amounts of DNA. Further, the minimum depth of coverage for whole-exome sequencing and whole-exome sequencing can vary with a median range between 30 × and 150 ×.45 Therefore, the detection of genetic mutations with a low mutational allelic frequency, such as those involving TP53 in esophageal epidermoid metaplasia specimens, by whole-genome sequencing and whole-exome sequencing is unlikely. In comparison, the minimum depth of coverage for targeted next-generation sequencing performed herein was between 500 × and 1000 × for each genomic region.
In summary, esophageal epidermoid metaplasia is a pathologic condition that we believe is a precursor lesion to esophageal squamous neoplasia. Although likely rare, esophageal epidermoid metaplasia may also be overlooked based on a general lack of awareness of its existence as a diagnostic entity. Consequently, recognition of esophageal epidermoid metaplasia by pathologists and gastroenterologists alike is imperative in developing appropriate surveillance protocols. Moreover, the detection of TP53 mutations in esophageal epidermoid metaplasia specimens may serve as an early detection biomarker for high-grade squamous dysplasia/esophageal squamous cell carcinoma.
References
Pennathur A, Gibson MK, Jobe BA et al, Oesophageal carcinoma. Lancet 2013;381:400–412.
Ferlay J, Soerjomataram I, Dikshit R et al, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–E386.
Enzinger PC, Mayer RJ . Esophageal cancer. N Engl J Med 2003;349:2241–2252.
Pennathur A, Farkas A, Krasinskas AM et al, Esophagectomy for T1 esophageal cancer: outcomes in 100 patients and implications for endoscopic therapy. Ann Thorac Surg 2009;87:1048–1054.
Ohashi S, Miyamoto S, Kikuchi O et al, Recent advances from basic and clinical studies of esophageal squamous cell carcinoma. Gastroenterology 2015;149:1700–1715.
Dawsey SM, Lewin KJ, Wang GQ et al, Squamous esophageal histology and subsequent risk of squamous cell carcinoma of the esophagus. A prospective follow-up study from Linxian, China. Cancer 1994;74:1686–1692.
Wang GQ, Abnet CC, Shen Q et al, Histological precursors of oesophageal squamous cell carcinoma: results from a 13 year prospective follow up study in a high risk population. Gut 2005;54:187–192.
Taylor PR, Abnet CC, Dawsey SM . Squamous dysplasia–the precursor lesion for esophageal squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev 2013;22:540–552.
Singhi AD, Arnold CA, Crowder CD et al, Esophageal leukoplakia or epidermoid metaplasia: a clinicopathological study of 18 patients. Mod Pathol 2014;27:38–43.
Ezoe Y, Fujii S, Muto M et al, Epidermoid metaplasia of the esophagus: endoscopic feature and differential diagnosis. Hepatogastroenterology 2011;58:809–813.
Fukui T, Sakurai T, Miyamoto S et al, Education and imaging. Gastrointestinal: epidermal metaplasia of the esophagus. J Gastroenterol Hepatol 2006;21:1627.
Kaye MD . Esophageal leukoplakia. Gastrointest Endosc 1987;33:254–259.
Nakanishi Y, Ochiai A, Shimoda T et al, Epidermization in the esophageal mucosa: unusual epithelial changes clearly detected by Lugol's staining. Am J Surg Pathol 1997;21:605–609.
Taggart MW, Rashid A, Ross WA et al, Oesophageal hyperkeratosis: clinicopathological associations. Histopathology 2013;63:463–473.
Cottreau J, Gruchy S, Kamionek M et al, Prevalence of oesophageal epidermoid metaplasia in 1048 consecutive patients and 58 patients with squamous neoplasms. Histopathology 2016;68:988–995.
Abedi-Ardekani B, Kamangar F, Sotoudeh M et al, Extremely high Tp53 mutation load in esophageal squamous cell carcinoma in Golestan Province, Iran. PLoS ONE 2011;6:e29488.
Xing EP, Nie Y, Song Y et al, Mechanisms of inactivation of p14ARF, p15INK4b, and p16INK4a genes in human esophageal squamous cell carcinoma. Clin Cancer Res 1999;5:2704–2713.
Agrawal N, Jiao Y, Bettegowda C et al, Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov 2012;2:899–905.
Lin DC, Hao JJ, Nagata Y et al, Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet 2014;46:467–473.
Song Y, Li L, Ou Y et al, Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014;509:91–95.
Zhang L, Zhou Y, Cheng C et al, Genomic analyses reveal mutational signatures and frequently altered genes in esophageal squamous cell carcinoma. Am J Hum Genet 2015;96:597–611.
Gao YB, Chen ZL, Li JG et al, Genetic landscape of esophageal squamous cell carcinoma. Nat Genet 2014;46:1097–1102.
Nikiforova MN, Wald AI, Melan MA et al, Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol 2016;18:379–387.
Jennings L, Van Deerlin VM, Gulley ML et al, Recommended principles and practices for validating clinical molecular pathology tests. Arch Pathol Lab Med 2009;133:743–755.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
Ng PC, Henikoff S . SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814.
Adzhubei IA, Schmidt S, Peshkin L et al, A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249.
Sherry ST, Ward MH, Kholodov M et al, dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311.
Forbes SA, Beare D, Gunasekaran P et al, COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811.
Grasso C, Butler T, Rhodes K et al, Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn 2015;17:53–63.
Dacic S, Kothmaier H, Land S et al, Prognostic significance of p16/cdkn2a loss in pleural malignant mesotheliomas. Virchows Arch 2008;453:627–635.
Salaria SN, Abu Alfa AK, Cruise MW et al, Lichenoid esophagitis: clinicopathologic overlap with established esophageal lichen planus. Am J Surg Pathol 2013;37:1889–1894.
Casson AG, Tammemagi M, Eskandarian S et al, p53 alterations in oesophageal cancer: association with clinicopathological features, risk factors, and survival. Mol Pathol 1998;51:71–79.
Egashira A, Morita M, Kakeji Y et al, p53 gene mutations in esophageal squamous cell carcinoma and their relevance to etiology and pathogenesis: results in Japan and comparisons with other countries. Cancer Sci 2007;98:1152–1156.
Vijeyasingam R, Darnton SJ, Jenner K et al, Expression of p53 protein in oesophageal carcinoma: clinicopathological correlation and prognostic significance. Br J Surg 1994;81:1623–1626.
Shi ST, Yang GY, Wang LD et al, Role of p53 gene mutations in human esophageal carcinogenesis: results from immunohistochemical and mutation analyses of carcinomas and nearby non-cancerous lesions. Carcinogenesis 1999;20:591–597.
Wang LD, Zhou Q, Hong JY et al, p53 protein accumulation and gene mutations in multifocal esophageal precancerous lesions from symptom free subjects in a high incidence area for esophageal carcinoma in Henan, China. Cancer 1996;77:1244–1249.
Slaughter DP, Southwick HW, Smejkal W . Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953;6:963–968.
Strong MS, Incze J, Vaughan CW . Field cancerization in the aerodigestive tract—its etiology, manifestation, and significance. J Otolaryngol 1984;13:1–6.
Regad T . Targeting RTK signaling pathways in cancer. Cancers 2015;7:1758–1784.
Hanawa M, Suzuki S, Dobashi Y et al, EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int J Cancer 2006;118:1173–1180.
Sunpaweravong P, Sunpaweravong S, Puttawibul P et al, Epidermal growth factor receptor and cyclin D1 are independently amplified and overexpressed in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol 2005;131:111–119.
Kobayashi H, Kumagai K, Gotoh A et al, Upregulation of epidermal growth factor receptor 4 in oral leukoplakia. Int J Oral Sci 2013;5:14–20.
Jyothi Meka N, Ugrappa S, Velpula N et al, Quantitative immunoexpression of EGFR in oral potentially malignant disorders: oral leukoplakia and oral submucous fibrosis. J Dent Res Dent Clin Dent Prospects 2015;9:166–174.
Sims D, Sudbery I, Ilott NE et al, Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 2014;15:121–132.
Acknowledgements
We would like to thank Ms Katelyn Smith for outstanding administrative assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Singhi, A., Arnold, C., Lam-Himlin, D. et al. Targeted next-generation sequencing supports epidermoid metaplasia of the esophagus as a precursor to esophageal squamous neoplasia. Mod Pathol 30, 1613–1621 (2017). https://doi.org/10.1038/modpathol.2017.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.73
