
Abstract
The distinction between chronic eosinophilic leukemia, not otherwise specified and idiopathic hypereosinophilic syndrome largely relies on clonality assessment. Prior to the advent of next-generation sequencing, clonality was usually determined by cytogenetic analysis. We applied targeted next-generation sequencing panels designed for myeloid neoplasms to bone marrow specimens from a cohort of idiopathic hypereosinophilic syndrome patients (n=51), and assessed the significance of mutations in conjunction with clinicopathological features. The findings were further compared with those of 17 chronic eosinophilic leukemia, not otherwise specified patients defined by their abnormal cytogenetics and/or increased blasts. Mutations were detected in 14/51 idiopathic hypereosinophilic syndrome patients (idiopathic hypereosinophilic syndrome/next-generation sequencing-positive) (28%), involving single gene in 7 and ≥2 in 7 patients. The more frequently mutated genes included ASXL1 (43%), TET2 (36%), EZH2 (29%), SETBP1 (22%), CBL (14%), and NOTCH1 (14%). Idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients showed a number of clinical features and bone marrow findings resembling chronic eosinophilic leukemia, not otherwise specified. Chronic eosinophilic leukemia, not otherwise specified patients showed a disease-specific survival of 14.4 months, markedly inferior to idiopathic hypereosinophilic syndrome/next-generation sequencing-negative (P<0.001), but not significantly different from idiopathic hypereosinophilic syndrome/next-generation sequencing-positive (P=0.117). These data suggest that targeted next-generation sequencing helps to establish clonality in a subset of patients with hypereosinophilia that would otherwise be classified as idiopathic hypereosinophilic syndrome. In conjunction with other diagnostic features, mutation data can be used to establish a diagnosis of chronic eosinophilic leukemia, not otherwise specified in patients presenting with hypereosinophilia.
Similar content being viewed by others

Introduction
Hypereosinophilia is defined as persistent and marked eosinophilia greater than 1.5 × 109/l. The diagnostic work-up of patients presenting with hypereosinophilia is challenging and requires resource-intensive testing. Primary eosinophilia(s) associated with translocation of platelet-derived growth factor receptor alpha (PDGFRA) and PDGFRbeta (PDGFRB), or fibroblast growth factor receptor 1 (FGFR1) have been recognized as genetically defined entities in the 2008 World Health Organization classification scheme of myeloid neoplasms.1 Chronic eosinophilic leukemia, not otherwise specified2 is a clonal hematological malignancy, currently classified within the group of myeloproliferative neoplasms without the presence of PDGFRA, PDGFRB, or FGFR1 abnormalities. Idiopathic hypereosinophilic syndrome, on the other hand, is defined by the presence of persistent, unexplained hypereosinophilia with associated organ damage without a known underlying cause and a lack of identifiable genetic abnormalities. Chronic eosinophilic leukemia, not otherwise specified and idiopathic hypereosinophilic syndrome are diagnoses of exclusion, which not only require exclusion of a known reactive process, genetically defined eosinophilic neoplasms, but also hypereosinophilia associated with a known myeloid neoplasm. The latter includes acute myeloid leukemia, myelodysplastic syndrome (MDS), systemic mastocytosis, chronic myeloid leukemia, other types of classical myeloproliferative neoplasm (essential thrombocythemia, polycythemia vera, and primary myelofibrosis), and chronic myelomonocytic leukemia; also excluded are cases of hypereosinophilia associated with a clonal abnormal T-cell population, so-called ‘lymphocytic variant’ hypereosinophilic syndrome.
Chronic eosinophilic leukemia, not otherwise specified is distinguished from idiopathic hypereosinophilic syndrome by the demonstration of clonality or increased blasts according to the 2008 World Health Organization Classification criteria. Until recently, clonality in clinical practice has been largely assessed by conventional karyotyping and molecular testing for the well-defined mutations associated with myeloproliferative neoplasm, such as KIT and JAK2 V617F. Hypereosinophilia in which reactive causes have been excluded, but lack of a definite cytogenetic/genetic abnormality, are currently lumped together under an umbrella term of idiopathic hypereosinophilic syndrome. As a result, idiopathic hypereosinophilic syndrome has become a heterogeneous entity with variable clinical behavior in affected patients.3, 4
In recent years, next-generation sequencing has helped to identify mutations in a large proportion of cases of myeloid neoplasms. In myeloproliferative neoplasms, discoveries include CALR mutations in JAK2 V617-negative classic myeloproliferative neoplasms,5, 6 as well as a number of recurrent mutations that correlate with the clinical features, prognosis, and treatment responses.7, 8, 9 Mutational analysis in general can help to differentiate a clonal hematopoietic neoplasm from a reactive process in diagnostically challenging cases. However, this approach has been complicated by reports of frequent somatic mutations in healthy populations of older individuals.10, 11, 12 The aim of this multicenter study was to molecularly characterize the cases of idiopathic hypereosinophilic syndrome, and compare them with chronic eosinophilic leukemia, not otherwise specified, as defined by the current World Health Organization criteria. Specifically, we sought to determine whether mutation data would provide clinically meaningful information in these two entities.
Materials and methods
Patients
We searched the pathology archives at seven institutions in the United States: MD Anderson Cancer Center, Stanford University Medical Center, Cleveland Clinic, Massachusetts General Hospital, Weill Cornell Medical College, the Hospital of the University of Pennsylvania, and the University of New Mexico for cases of persistent hypereosinophilia between 2005 and 2014. All included cases had to meet the minimal 2008 World Health Organization Classification requirements for persistent hypereosinophilia (≥1.5 × 109/l). Chronic eosinophilic leukemia, not otherwise specified was characterized either by an increase in blasts (≥5%) in the bone marrow or blood (≥2%) and/or a clonal cytogenetic abnormality, after excluding other myeloid neoplasms that might be associated with eosinophilia (acute leukemias, chronic myeloid leukemia, other defined myeloproliferative neoplasm subtypes, MDS, chronic myelomonocytic leukemia, systemic mastocytosis, or cases with recurrent PDGFRA/B or FGFR1 rearrangements). Idiopathic hypereosinophilic syndrome was defined as persistent hypereosinophilia with end-organ damage, with no clear causes for a primary or secondary cause of hypereosinophilia. The definition of 'end-organ damage' was according to the proposals by the working group on eosinophil disorders and syndromes.13 Cases of so-called lymphocytic/T-cell variant hypereosinophilic syndrome,3 which showed aberrant T cells immunophenotypically by flow cytometry, were excluded. Clinical information was retrieved from the electronic medical records. This study was approved by the Institutional Review Boards of all participating institutions.
Cytogenetics, Fluorescence in situ Hybridization, and Molecular Testing
Conventional cytogenetic analysis was performed on G-banded metaphase cells prepared from unstimulated bone marrow aspirate cultures using standard techniques. Twenty metaphases were analyzed and the results were reported using the International System for Human Cytogenetic Nomenclature. Fluorescence in situ hybridization and/or molecular genetic methods for detecting BCR-ABL1, PDGFRA, PDGFRB, or FGFR1 were performed at respective institutions as part of the routine clinical work-up.
Bone Marrow Morphologic and Histologic Assessment
The evaluated histological criteria were agreed by all participants prior to case collection to ensure consistency in morphological assessment. The bone marrow aspirate smears and core biopsy either obtained at the time of diagnosis or the first bone marrow procedure ever performed with paired peripheral blood smears were reviewed by at least one observer at each institution. Bone marrow fibrosis was assessed by reticulin and trichrome stains and graded according to the European bone marrow Fibrosis Network criteria.14 Megakaryocyte morphology was recorded as predominantly MDS-like (small with hypolobated nuclei or abnormally lobated nuclei), predominantly myeloproliferative neoplasm-like (large and hypersegmented megakaryocytes, often with clustering), mixed myelodysplastic and myeloproliferative neoplasm-like, or within normal limits. CD2 and CD25, either by flow cytometry or by immunohistochemistry or both, were assessed in cases with increased mast cells. Cases that met the diagnostic criteria of systemic mastocytosis were excluded. A differential count based on at least 200 cells (or all available cells if <200 present) was performed, and ring sideroblasts were assessed on Prussian blue or Perls-stained bone marrow aspirate smears. For eosinophil morphology, abnormal features included abnormal granulation (hypogranulation or uneven granulation), cytoplasmic vacuoles, and abnormal nuclear lobation (monolobated or multinucleated). Normal eosinophils have bilobed nuclei and evenly distributed eosinophilic granules. Mild nuclear hypersegmentation and mild hyper, hypo, or uneven granulation of eosinophils were not considered to be abnormal because these changes can be associated with eosinophil activation15 or in patients treated with hydroxyurea.16 Cases in which the submitter was uncertain of the diagnosis and/or specific histologic features were reviewed together using a multi-headed microscope by six of the authors (SW, AO, DA, RH, EH, and AB). All histological review was blinded to next-generation sequencing testing results.
Targeted Next-Generation Sequencing
We performed targeted next-generation sequencing on DNA samples extracted from frozen unfractionated bone marrow cells collected at the time of diagnosis. In a subset of cases, DNA was extracted from unstained and unfixed bone marrow aspirate smears. Sequences were performed with using a next-generation sequencing-based custom-designed assay using the Illumina MiSeq platform. The entire coding sequences of 45 genes, including ABL1, ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, DNMT3A, ETV6, EZH2, FAM5C, FLT3-ITD, FLT3-TKD, GATA1, GATA2, HNRNPK, IDH1, IDH2, IKZF1, JAK1, JAK2, KDM6A, KIT, KRAS, MPL, NFE2, NOTCH1, NPM1, NRAS, PHF6, PTPN11, RAD21, RUNX1, SEPBP1, SF3B1, SH2B3, SMC1A, SMC3, STAG2, SUZ12, TET2, TP53, U2AF1, WT1, and ZRSR2, were performed specifically for this study. A few cases were performed with a 28-gene panel sequencing the entire coding regions, or a 53-gene hot-spot sequencing panel at local hospitals as part of the clinical work-up. The 28-gene8 panel included ABL1, ASXL1, BRAF, DNMT3A, EGFR, EZH2, FLT3, GATA1, GATA2, HRAS, IDH1, IDH2, KIT, KRAS, MDM2, IKZF2, JAK2, KMT2A, MPL, MYD88, NOTCH1, NPM1, NRAS, PTPN11, RUNX1, TET2, TP53, and WT1, with CALR, FLT3, and CEBPA tested separately by Sanger sequencing. The 53-gene panel17 included ASXL1, BCOR, BCORL1, BRAF, BRINP3, CALR, CBL, CEBPA, CSF3R, DNMT1, DNMT3A, EED, ETV6, EZH2, FLT3, GATA1, GATA2, HNRNPK, IDH1, IDH2, JAK2, JAK3, KDM6A, KIT, KMT2A, KRAS, LUC7L2, MPL, NOTCH1, NPM1, NRAS, NSD1, PHF6, PRPF40B, PTPN11, RAD21, RUNX1, SETBP1, SF1, SF3A1, SF3B1, SMC1A, SMC3, SRSF2, STAG2, SUZ12, TET1, TET2, TP53, U2AF1, U2AF2, WT1, and ZRSR2. Variant calling was performed with Illumina MiSeq Reporter Software 1.3.17. using human genome build 19 (hg 19) as a reference. We added this to method.
Statistical Analyses
For continuous variables, data are reported as median and range. For nominal variables, data are reported as the number of patients unless otherwise specified. Disease-specific survival was calculated from the day of diagnosis to the last follow-up or death attributed to chronic eosinophilic leukemia, not otherwise specified/idiopathic hypereosinophilic syndrome. Patients who received hematopoietic stem cell transplant were censored at the time of the procedure. Distribution of disease-specific survival was estimated by Kaplan–Meier curves. Fisher’s exact and Chi-square tests were used for categorical comparisons. All P values are two-tailed and were considered significant when <0.05. No adjustments for multiplicity were made.
Results
Patients and Mutational Studies
From an initial collection of patients with hypereosinophilia, we reviewed the clinical presentation, laboratory data and bone marrow findings and identified 125 cases meeting the diagnostic criteria for idiopathic hypereosinophilic syndrome or chronic eosinophilic leukemia, not otherwise specified. PDGFRA/B or FGFR1 rearrangements were confirmed to be negative by cytogenetics/fluorescence in situ hybridization and/or molecular genetic methods in all cases. Clonal cytogenetic abnormalities were detected in 16 patients. Of those, four patients had a complex karyotype, one had two karyotypic abnormalities, nine had a single abnormality, and one was identified by fluorescence in situ hybridization to have del(9p) abnormality (Supplementary Table 1). A total of 57 patients with material available were tested by next-generation sequencing, including 6 chronic eosinophilic leukemia, not otherwise specified patients (5 patients with cytogenetic abnormalities and 1 patient with a normal karyotype but increased blasts) and 51 idiopathic hypereosinophilic syndrome. Of those, 46 patients had DNA samples extracted from fresh or frozen cells, 11 from bone marrow aspirate smears (n=11), and 9 patients had both. The latter nine cases showed excellent test concordance between the two types of archived samples. The 45-gene panel (sequencing >90% gene coding regions) was conducted on 44 patient samples, 28-gene panel (sequencing entire coding regions) on 9, and 50-gene panel on 4. Among the nine samples tested by the 28-gene panel, CSF3R mutation was additionally tested by Sanger sequencing in two patients.
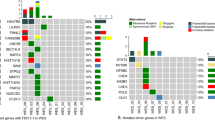
Pathologic mutations (allele burden ≥5%) were detected in 3/6 (50%) chronic eosinophilic leukemia, not otherwise specified patients, and 14/51 (28%) idiopathic hypereosinophilic syndrome patients (Table 1). Of the 14 idiopathic hypereosinophilic syndrome patients with mutations, 7 (50%) had one mutation, 5 (36%) had two mutations and 2 (14%) had three or more mutations (14%). Involved genes, in an order of decreasing frequency, were: ASXL1 (6/14, 43%), TET2 (5/14, 36%), EZH2 (4/14, 29%), SETBP1 (2/9, 22%), CBL (2/14, 14%), NOTCH1 (2/14, 14%), and 1 (7%) each of DNMT3A, NRAS, JAK2 exon 13, and GATA2. The allele frequency is shown in Supplementary Table 1. Mutations that characterize classic myeloproliferative neoplasms, including JAK2 V617F (0/51), MPL (0/51), and CALR (0/45), were all negative. KIT mutations were also not detected in any of the cases (0/51). Of the two chronic eosinophilic leukemia, not otherwise specified patients with an abnormal karyotype, the patient with a complex karyotype showed a TP53 mutation and another patient with interstitial deletion of 5q had a GATA2 mutation. The other chronic eosinophilic leukemia, not otherwise specified patient who had a normal karyotype but increased blasts showed multiple mutations, including ASXL1, CSF3R, SETBP1, U2AF1, EZH2, and ETV6. The detailed information of nucleotide alterations and resultant amino acid changes are shown in Supplementary Table 2.
Clinical Features of Patients with a Normal Karyotype but Positive for Mutations
According to the current World Health Organization classification criteria, patients with clonal cytogenetic abnormalities and/or increased blasts in bone marrow or peripheral blood were grouped together as chronic eosinophilic leukemia, not otherwise specified (n=17). The remaining 51 patients who would otherwise be classified as idiopathic hypereosinophilic syndrome were further grouped by the presence (idiopathic hypereosinophilic syndrome/next-generation sequencing-positive) (n=14) or absence of mutations (idiopathic hypereosinophilic syndrome/next-generation sequencing-negative) (n=37).
The median age of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients were 63.8 years (24.3–89.0), older than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients (41.0 years, P<0.001) but comparable with chronic eosinophilic leukemia, not otherwise specified patients (66.4 years, P=0.660). Although WBC, eosinophil percentage, and absolute eosinophil count showed no differences among the three groups of patients, idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients, like chronic eosinophilic leukemia, not otherwise specified patients, more frequently presented with anemia (P=0.048) than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients. The platelet counts, if compared as continuous variables, showed no statistical significance among three groups of patients; however; when platelets were categorized as thrombocytosis (≥450 × 109/l), normal (140–450 × 109/l), and thrombocytopenia (<140 × 109/l), an abnormal platelet count was more frequently observed in chronic eosinophilic leukemia, not otherwise specified (P=0.009); borderline in idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients (P=0.081), compared with idiopathic hypereosinophilic syndrome/next-generation sequencing-negative. Of note, chronic eosinophilic leukemia, not otherwise specified patients appeared to show predominantly thrombocytopenia, whereas idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients showed either thrombocytosis (n=4) or thrombocytopenia (n=3).
Regarding clinical features, compared with idiopathic hypereosinophilic syndrome/next-generation sequencing-negative, chronic eosinophilic leukemia, not otherwise specified patients presented with more constitutional symptoms (weight loss, night sweats, and progressive weakness) or cytopenia(s)-related symptoms (infection or anemia-associated symptoms); but less eosinophil-mediated symptoms such as allergy/skin lesions, edema, rheumatoid symptoms, or hypersensitivity. Chronic eosinophilic leukemia, not otherwise specified patients frequently had a high lactate dehydrogenase (P=0.005), and borderline more frequent organomegaly (P=0.066). The clinical characteristics of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients varied between chronic eosinophilic leukemia, not otherwise specified and idiopathic hypereosinophilic syndrome/next-generation sequencing-negative (Tables 1 and 2). As a group, similar to idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients, idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients showed more frequent eosinophilic activation-related symptoms, less organomegaly, or elevated lactate dehydrogenase than chronic eosinophilic leukemia, not otherwise specified; on the other hand, like chronic eosinophilic leukemia, not otherwise specified, idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients were older, with more frequent anemia, and showed a trend for more frequent constitutional or cytopenia-related symptoms, contrasting to idiopathic hypereosinophilic syndrome/next-generation sequencing-negative.
Bone Marrow Morphologic and Histological Findings
Essentially all chronic eosinophilic leukemia, not otherwise specified patients showed an age-adjusted bone marrow hypercellularity (Figure 1a), and 11/17 (65%) patients had a bone marrow cellularity ≥90%, significantly higher than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients (P<0.001) as well as idiopathic hypereosinophilic syndrome/next-generation sequencing-positive (P=0.024) patients (Table 2). Bone marrow blasts ≥5% were seen in three chronic eosinophilic leukemia, not otherwise specified patients. Increased bone marrow eosinophils (median 20%, range 5–91%) and increased myeloid:erythroid ratio (4.8, range 1.3–16) (normal range 2:1–4:1) on the other hand, were similarly observed in all three groups of patients. Regarding megakaryocyte morphology, typical myeloproliferative neoplasm-type megakaryocytes were very uncommon, being seen in only one chronic eosinophilic leukemia, not otherwise specified patient, whereas MDS-like megakaryocytes (Figure 1a) or a mixed MDS and myeloproliferative neoplasm-like megakaryocyte morphology were frequently observed in chronic eosinophilic leukemia, not otherwise specified, compared with idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients (P<0.001). Significant dyserythropoiesis (Figure 1b) and dysgranulopoiesis or both were seen in 6/17 chronic eosinophilic leukemia, not otherwise specified patients (P=0.003), significantly more frequent than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients. Similar to chronic eosinophilic leukemia, not otherwise specified, idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients also showed more frequent abnormal megakaryocytes (P=0.009) as well as dysgranulopoiesis and/or dyserythropoiesis (P=0.017). Overall, very few patients in the group of idiopathic hypereosinophilic syndrome/next-generation sequencing-negative showed abnormal megakaryocytes, dyserythropoiesis, and dysgranulopoiesis. Eosinophil morphology was assessed both on bone marrow aspirate smears and peripheral blood smears. Abnormal eosinophil granulation and abnormal nuclear lobation (multilobation, hypolobation, abnormal nuclear branching, but not considering mild hypersegmentation, Figure 1b) were seen in all three groups of patients, but were significantly more frequent in chronic eosinophilic leukemia, not otherwise specified (P=0.010) and also showed a trend for a higher frequency in idiopathic hypereosinophilic syndrome/next-generation sequencing-positive (P=0.078) compared with idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients. MF-1 fibrosis was observed in 28% and MF2/MF3 fibrosis in 10% of all patients, showing no statistical significance among the three groups. A slight increase in mast cells, mostly in an interstitial pattern, was observed in five patients of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive, two of idiopathic hypereosinophilic syndrome/next-generation sequencing-negative, and one chronic eosinophilic leukemia, not otherwise specified (not significant). Notably, none of these cases showed KIT mutation or immunophenotypic aberrancies in mast cells, and none met the criteria for systemic mastocytosis.

Histological abnormalities frequently observed in chronic eosinophilic leukemia, not otherwise specified and with mutations. (a) Bone marrow hypercellularity with small hypolobated megakaryocytes (arrows) (hemotoxylin and eosin, original magnification, × 500); (b) Numerous eosinophils with abnormal granulation and abnormal nuclear segmentation and dyserythropoiesis (arrow) (Wright-Giemsa, original magnification, × 1000).
Treatment and Clinical Outcomes
The treatment was non-uniform and included various agents that were often used sequentially over the course of disease for all three groups of patients. These included hydroxyurea (for cytoreduction), corticosteroids with or without interferon, cyclosporine, methotrexate, and alemtuzumab. Tyrosine kinase inhibitors, mostly imatinib and occasionally dasatinib, were used in 28/66 (42%) patients. Hypomethylating agents, single agent chemotherapy, and high-dose chemotherapy were also used in some patients when disease showed progression or was refractory to other treatment modalities. Four patients received hematopoietic stem cell transplant (Supplementary Table 2). Compared with chronic eosinophilic leukemia, not otherwise specified, idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients were treated more often with corticosteroids (28/37 vs 7/16, P=0.056) and fewer patients were treated with cytoreduction (8/37 vs 8/15, P=0.045) at initial treatment; and fewer patients required a hypomethylating agent or chemotherapy in the course of disease (P=0.021). The treatment modalities administered to idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients were essentially the same as those given to chronic eosinophilic leukemia, not otherwise specified patients, but showed a trend for more hydroxyurea (P=0.083) and hypomethylating agent/chemotherapy (P=0.058) than idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients (Supplementary Table 3). Other treatment modalities were not significantly different among the three groups, including the use of tyrosine kinase inhibitor.
The median follow-up time, including dead and living patients, was 24.9 months (range 0.6–405.3 months). In the chronic eosinophilic leukemia, not otherwise specified group, two patients progressed to acute myeloid leukemia and one progressed with increased bone marrow blasts from 4% to 17%. None of the patients with idiopathic hypereosinophilic syndrome/next-generation sequencing-positive or idiopathic hypereosinophilic syndrome/next-generation sequencing-negative showed progression to acute myeloid leukemia. During the follow-up, three patients died of unrelated causes, including colon cancer, diffuse large B-cell lymphoma, and accidental death; therefore, the outcome was compared with disease-specific survival. The median disease-specific survival of chronic eosinophilic leukemia, not otherwise specified patients was 14.4 months (range 1.0–120.1 months), whereas it was not reached for idiopathic hypereosinophilic syndrome/next-generation sequencing-negative or idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients. By Kaplan–Meier log rank test, the disease-specific survival of chronic eosinophilic leukemia, not otherwise specified patients was significantly inferior to idiopathic hypereosinophilic syndrome/next-generation sequencing-negative (P<0.001), but not significantly different from idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients (P=0.117). The disease-specific survival of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive was shorter than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients, but this did not reach statistical significance (P=0.105) (Figure 2).

Patients with chronic eosinophilic leukemia, not otherwise specified showed a median disease-specific survival of 14.4 months (1.0–120.1 months), significantly inferior to patients with idiopathic hypereosinophilic syndrome with no identifiable mutations (median disease-specific survival: not reached, 0.6–405.3 months, P<0.001), but not statistically different from idiopathic hypereosinophilic syndrome patients with mutations (not reached, 1.0–223.9 months, P=0.117).
Discussion
Using targeted sequencing panels designed for myeloid neoplasms, we found somatic mutations in 28% of patients with a diagnosis and classification of idiopathic hypereosinophilic syndrome according to current World Health Organization criteria that excluded lymphocyte variant hypereosinophilic syndrome and hypereosinophilic syndrome associated with recurrent genetic abnormalities. Mutations frequently affected genes involving DNA methylation and chromatin modification. Idiopathic hypereosinophilic syndrome patients with proven mutations, as a group, showed a number of clinical, laboratory, and bone marrow features resembling chronic eosinophilic leukemia, not otherwise specified.
The next-generation sequencing panels we used in this study included the most frequently found mutated genes in myeloid neoplasms, including genes encoding signaling molecules, transcription factors, epigenetic regulators, and splicing factors. Using these panels, we found that, except for one case that showed a JAK2 exon 13 mutation, mutations of classic myeloproliferative neoplasms such as JAK2 V617F, MPL, and CALR, were not present in our idiopathic hypereosinophilic syndrome/chronic eosinophilic leukemia, not otherwise specified patients. KIT mutations, including KIT 816 V and KIT M541L, were not found either. KIT M541L mutations were recently reported in four of five chronic eosinophilic leukemia, not otherwise specified patients who did not have PDGFRA/B lesions but responded to tyrosine kinase inhibitor treatment.18 Schwaab and colleagues19 reported KIT 816 V mutations in 3% (14 patients) and JAK2 V617F mutations in 4% (17 patients) of patients who presented with hypereosinophilia of unknown significance. Notably, with the mutation information and upon review of the slides, the authors reclassified the cases with KIT D816V as systemic mastocytosis with eosinophilia (systemic mastocytosis-eo) and cases with JAK2 V617F mutation as myeloproliferative neoplasm with eosinophilia (myeloproliferative neoplasm-eo). In our study, the bone marrow slides and most peripheral blood smears were reviewed by experienced hematopathologists in conjunction with clinical, laboratory, and radiographic data, prior to the mutation study. We had a small subset of patients showing a mild increase in mast cells in an interstitial pattern in bone marrow; however, none of them met the current diagnostic criteria for systemic mastocytosis. The most frequent mutations found in our idiopathic hypereosinophilic syndrome patients were ASXL1, TET2, and EZH2, genes involved in DNA methylation and chromatin modification.20, 21 SETBP1 and CSF3R, tested in a subset of our idiopathic hypereosinophilic syndrome and chronic eosinophilic leukemia, not otherwise specified cases, were also found to be mutated in some patients. SETBP1 has been found frequently mutated in chronic neutrophilic leukemia,22 atypical chronic myeloid leukemia, myelodysplastic/myeloproliferative neoplasm-unclassifiable, and chronic myelomonocytic leukemia,23 whereas CSF3R mutation is highly prevalent in chronic neutrophilic leukemia but appears to be less common in atypical chronic myeloid leukemia.24, 25 These two genes will be of great interest to investigate further, because the clinical and pathological features of chronic eosinophilic leukemia, not otherwise specified in our study closely resembled non-classic myeloproliferative neoplasm or MDS/myeloproliferative neoplasm overlap disease. Recently, Anderson and colleagues26 isolated eosinophils and performed whole genome sequencing in five patients with idiopathic hypereosinophilic syndrome. Somatic missense mutations were found in three patients, including spliceosome gene PUF60 and the cadherin gene CDH17. These genes were not included in our panel and their prevalence and significance in idiopathic hypereosinophilic syndrome require further investigation. They further showed that reactive eosinophilia samples could be differentiated from known and suspected clonal eosinophilias based on 285 differentially methylated CpG sites corresponding to 128 differentially methylated genes.
The second question we tried to address is whether finding mutations is sufficient to diagnose a case as a clonal myeloid neoplasm that is distinctly different from other idiopathic hypereosinophilic syndrome. We first compared the clinical features and pathology findings of chronic eosinophilic leukemia, not otherwise specified with idiopathic hypereosinophilic syndrome with no identifiable mutations. Chronic eosinophilic leukemia, not otherwise specified patients were older and showed significantly more frequent constitutional symptoms, more anemia and thrombocytopenia, and symptoms related to cytopenia(s), frequent organomegaly, elevated lactate dehydrogenase; but less allergy, skin urticarial/rash, edema, asthma, myalgia/arthralgia, or eosinophil-mediated organ injury. These clinical features more closely resemble those of an aggressive myeloid neoplasm and were different from the clinical features seen in most of the idiopathic hypereosinophilic syndrome in our series. In our study, chronic eosinophilic leukemia, not otherwise specified patients had a median disease-specific survival of 14.4 months, and three cases progressed to acute myeloid leukemia or accelerated phase. Our findings are similar to those reported by Helbig and colleagues27 and suggest that chronic eosinophilic leukemia, not otherwise specified is a clinically aggressive disease, often resistant to therapy, with a relatively high rate of acute myeloid leukemia transformation and a short survival. In contrast, idiopathic hypereosinophilic syndrome patients with no identifiable mutations presented with hypereosinophilia at a much younger age than chronic eosinophilic leukemia, not otherwise specified, and had more frequent symptoms associated with eosinophil activation, such as dermatological, pulmonary, gastrointestinal, and rheumatologic manifestations, as has been reported by others.4, 28
Compared with the contrasting clinical features of idiopathic hypereosinophilic syndrome/next-generation sequencing-negative vs chronic eosinophilic leukemia, not otherwise specified, the clinical manifestations of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive as a group were more heterogeneous, but showed some similarities to chronic eosinophilic leukemia, not otherwise specified. The disease-specific survival of chronic eosinophilic leukemia, not otherwise specified patients was significantly shorter than that of idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients (P<0.001), but was not statistically different from idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients (P=0.117). Although this may be due to the relatively small number of patients with mutations that limited the statistical power, several other explanations are also plausible. The next-generation sequencing assays used in this study targeted genes that are known to be mutated with relatively high frequency in myeloid neoplasms; although with good gene coverage, some relevant genes were either not included, such as above-mentioned PUF60 and CDH17,26 or only tested in a subset of patients, such as SETBP1 and CSF3R. We may have found mutations in idiopathic hypereosinophilic syndrome/next-generation sequencing-negative patients if whole genome sequencing or a more extensive next-generation sequencing panel had been performed. On the other hand, somatic mutations in genes associated with myeloid neoplasms (such as DNMT3A, TET2, JAK2, ASXL1, TP53, GNAS, PPM1D, BCORL1, and SF3B1) have been frequently found in aging healthy individuals.10, 11, 12, 29 MDS-associated somatic mutations and clonal hematopoiesis have been recently reported in near half of patients with aplastic anemia,30 as well as in patients with idiopathic cytopenias of undetermined significance.29, 31 In the latter, variant allele fractions were comparable with MDS. Although individuals with acquired somatic mutations may have increased risk for development of a myeloid neoplasm such as MDS or acute myeloid leukemia,32 the complex dynamics of clonal hematopoiesis are highly variable and are not necessarily predictable.33 In our cohort, idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients were significantly older than patients without mutations, and it is possible that some mutations, especially the ones with single mutation and a low allele frequency, were age-related. Certain specific mutations, mutations of high allele frequency, or a higher number of mutations may be more pathogenetically relevant. The small number of idiopathic hypereosinophilic syndrome cases with mutations in our study, however, precluded a meaningful comparison of individual mutations. On a further note, although an abnormal karyotype is highly associated with a neoplastic process, it has been recognized that not all clonal cytogenetic abnormities have equivalent diagnostic importance. Del(20q),34 +8, and +1535 have been recognized as clonal karyotypic abnormalities of uncertain significance in the absence of other diagnostic criteria for a myeloid neoplasm.36 Furthermore, by definition, cases with increased blasts, even in the absence of clonal cytogenetic abnormalities, were assigned to the chronic eosinophilic leukemia, not otherwise specified group, excluding these cases with aggressive features from the idiopathic hypereosinophilic syndrome group.
The morphological features of chronic eosinophilic leukemia, not otherwise specified and idiopathic hypereosinophilic syndrome are not well studied, likely owing to the rarity of these entities and unreliable features in distinguishing neoplastic vs reactive eosinophils.37, 38, 39, 40 The World Health Organization description of chronic eosinophilic leukemia, not otherwise specified2 was based on a few early studies,37, 38 and it is uncertain whether those early described cases included cases of PDGFRA/B, FGFR1, systemic mastocytosis-eo, or other myeloid neoplasms associated with hypereosinophilia. In our cohort, we found that abnormal megakaryocytes were seen in more than half of the chronic eosinophilic leukemia, not otherwise specified patients, showing frequent MDS-like or a mixture of MDS and myeloproliferative neoplasm-like features, whereas classic myeloproliferative neoplasm-like megakaryocytes were infrequent. Dysgranulopoiesis and dyserythropoiesis were also observed in a subset of our chronic eosinophilic leukemia, not otherwise specified cases. Reviewing the literature, although most of studies on CEL and idiopathic hypereosinophilic syndrome do not provide in detail histological descriptions, Weide et al41 and Kuk et al42 did describe multilineage dysplasia in their case report of chronic eosinophilic leukemia, not otherwise specified. Regarding the eosinophil morphology, we found frequent abnormalities in chronic eosinophilic leukemia, not otherwise specified and these were seen less frequently in idiopathic hypereosinophilic syndrome without mutations. Although the bone marrow and peripheral blood features of idiopathic hypereosinophilic syndrome/next-generation sequencing-positive patients were variable, they overall more closely resembled chronic eosinophilic leukemia, not otherwise specified than idiopathic hypereosinophilic syndrome/next-generation sequencing-negative. These findings indicate that molecular genetic events potentially important for leukemogenesis likely correlate with morphologic abnormalities; incorporating morphological assessment in the interpretation of molecular genetic data may help further identification of a true neoplasm from non-neoplastic hypereosinophilia.
In summary, we show in this study that myeloid neoplasm-related somatic mutations are present in a subset of eosinophilic proliferations that are currently classified as idiopathic hypereosinophilic syndrome. Mutations characteristics of classic myeloproliferative neoplasm, such as JAK2 V617F, MPL, and CALR, are infrequent. Rather, mutations are frequently found in genes involving DNA methylation and chromatin modification. Whereas idiopathic hypereosinophilic syndrome with no identifiable mutations has clinicopathological features that are distinct from chronic eosinophilic leukemia, not otherwise specified, idiopathic hypereosinophilic syndrome with mutations closely resembles chronic eosinophilic leukemia, not otherwise specified. Our findings indicate that cases of with mutations known to be associated with myeloid neoplasms should appropriately be classified as chronic eosinophilic leukemia, not otherwise specified. However, with the caveat of the high prevalence of mutations in aging populations, the detection of mutations needs to be interpreted in the context of other clinical and pathological findings when establishing a diagnosis of clonal hypereosinophilia.
References
Bain BJ, Gilliland DG, Horny H-P et al. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR1. In: Swerdlow SHCE, Campo E, Harris NL et al (eds). WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2008, pp 68–73.
Bain BJ, Gilliland DG, Vardiman JW et al. Chronic eosinophilic leukemia, not otherwise specified. In: Swerdlow SH, Campo E, Harris NL et al (eds). WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press, 2008, pp 51–53.
Gotlib J . World Health Organization-defined eosinophilic disorders: 2015 update on diagnosis, risk stratification, and management. Am J Hematol 2015;90:1077–1089.
Ogbogu PU, Bochner BS, Butterfield JH et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009; 124:1319–1325.e3.
Klampfl T, Gisslinger H, Harutyunyan AS et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–2390.
Nangalia J, Massie CE, Baxter EJ et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–2405.
Cazzola M, Kralovics R . From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood 2014;123:3714–3719.
Patel KP, Newberry KJ, Luthra R et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015;126:790–797.
Tenedini E, Bernardis I, Artusi V et al. Targeted cancer exome sequencing reveals recurrent mutations in myeloproliferative neoplasms. Leukemia 2014;28:1052–1059.
Jaiswal S, Fontanillas P, Flannick J et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–2498.
Genovese G, Kahler AK, Handsaker RE et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371:2477–2487.
Xie M, Lu C, Wang J et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014;20:1472–1478.
Valent P, Klion AD, Horny HP et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012;130:607–612.e9.
Thiele J, Kvasnicka HM, Facchetti F et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005;90:1128–1132.
Yamamoto A, Kojima T, Aoki T et al. Eosinophil hypersegmentation is a possible marker to monitor the disease activity of atopic dermatitis. Allergology International 2001;50:325–330.
Xu X . Nuclear hypersegmentation of neutrophils, eosinophils, and basophils due to hydroxycarbamide (hydroxyurea). Blood 2014;124:1392.
Shen W, Szankasi P, Sederberg M et al. Concurrent detection of targeted copy number variants and mutations using a myeloid malignancy next generation sequencing panel allows comprehensive genetic analysis using a single testing strategy. Br J Haematol 2016;173:49–58.
Iurlo A, Gianelli U, Beghini A et al. Identification of kit(M541L) somatic mutation in chronic eosinophilic leukemia, not otherwise specified and its implication in low-dose imatinib response. Oncotarget 2014;5:4665–4670.
Schwaab J, Umbach R, Metzgeroth G et al. KIT D816V and JAK2 V617F mutations are seen recurrently in hypereosinophilia of unknown significance. Am J Hematol 2015;90:774–777.
Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–2074.
Larsson CA, Cote G, Quintas-Cardama A . The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol Cancer Res 2013;11:815–827.
Cui Y, Li B, Gale RP et al. CSF3R, SETBP1 and CALR mutations in chronic neutrophilic leukemia. J. Hematol Oncol 2014;7:77.
Piazza R, Valletta S, Winkelmann N et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet 2013;45:18–24.
Pardanani A, Lasho TL, Laborde RR et al. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia 2013;27:1870–1873.
Wang SA, Hasserjian RP, Fox PS et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood 2014;123:2645–2651.
Andersen CL, Nielsen HM, Kristensen LS et al. Whole-exome sequencing and genome-wide methylation analyses identify novel disease associated mutations and methylation patterns in idiopathic hypereosinophilic syndrome. Oncotarget 2015;6:40588–40597.
Helbig G, Soja A, Bartkowska-Chrobok A et al. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am J Hematol 2012;87:643–645.
Fauci AS, Harley JB, Roberts WC et al. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 1982;97:78–92.
Malcovati L, Cazzola M . The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematology Am Soc Hematol Educ Program 2015;2015:299–307.
Yoshizato T, Dumitriu B, Hosokawa K et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med 2015;373:35–47.
Kwok B, Hall JM, Witte JS et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015;126:2355–2361.
Cargo CA, Rowbotham N, Evans PA et al. Targeted sequencing identifies patients with preclinical MDS at high risk of disease progression. Blood 2015;126:2362–2365.
Steensma DP, Bejar R, Jaiswal S et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015;126:9–16.
Yin CC, Peng J, Li Y et al. Clinical significance of newly emerged isolated del(20q) in patients following cytotoxic therapies. Mod Pathol 2015;28:1014–1022.
Goswami RS, Liang CS, Bueso-Ramos CE et al. Isolated +15 in bone marrow: disease-associated or a benign finding? Leuk Res 2015;39:72–76.
Vardiman JW, Thiele J, Arber DA et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937–951.
Chusid MJ, Dale DC, West BC et al. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975;54:1–27.
Flaum MA, Schooley RT, Fauci AS et al. A clinicopathologic correlation of the idiopathic hypereosinophilic syndrome. I. Hematologic manifestations. Blood 1981;58:1012–1020.
Kueck BD, Smith RE, Parkin J et al. Eosinophilic leukemia: a myeloproliferative disorder distinct from the hypereosinophilic syndrome. Hematol Pathol 1991;5:195–205.
Weller PF, Bubley GJ . The idiopathic hypereosinophilic syndrome. Blood 1994;83:2759–2779.
Weide R, Rieder H, Mehraein Y et al. Chronic eosinophilic leukaemia (CEL): a distinct myeloproliferative disease. Br J Haematol 1997;96:117–123.
Kuk JS, Maceachern JA, Soamboonsrup P et al. Chronic eosinophilic leukemia presenting with autoimmune hemolytic anemia and erythrophagocytosis by eosinophils. Am J Hematol 2006;81:458–461.
Acknowledgements
We thank Dr Duane Hassane at Weill Cornell Medical College, Bioinformatics Department in helping setting up the next-generation sequencing panel; Maitrayee Goswami at MD Anderson Cancer Center for her support in handling samples; Roxanna Anaya and Marisol Gonzalez at MD Anderson Cancer Center in coordinating IRB and MTA protocols.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This is a Bone Marrow Pathology Working Group Study
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, S., Tam, W., Tsai, A. et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod Pathol 29, 854–864 (2016). https://doi.org/10.1038/modpathol.2016.75
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.75
This article is cited by
-
Updates on eosinophilic disorders
Virchows Archiv (2023)
-
International Consensus Classification of myeloid and lymphoid neoplasms: myeloproliferative neoplasms
Virchows Archiv (2023)
-
Myeloid/lymphoid neoplasms with FLT3 rearrangement
Modern Pathology (2021)
-
Chronic myeloid neoplasms harboring concomitant mutations in myeloproliferative neoplasm driver genes (JAK2/MPL/CALR) and SF3B1
Modern Pathology (2021)
-
Myelodysplastic syndrome with t(6;9)(p22;q34.1)/DEK-NUP214 better classified as acute myeloid leukemia? A multicenter study of 107 cases
Modern Pathology (2021)
