
Abstract
Hereditary endometrial carcinoma is associated with germline mutations in Lynch syndrome genes. The role of other cancer predisposition genes is unclear. We aimed to determine the prevalence of cancer predisposition gene mutations in an unselected endometrial carcinoma patient cohort. Mutations in 25 genes were identified using a next-generation sequencing-based panel applied in 381 endometrial carcinoma patients who had undergone tumor testing to screen for Lynch syndrome. Thirty-five patients (9.2%) had a deleterious mutation: 22 (5.8%) in Lynch syndrome genes (three MLH1, five MSH2, two EPCAM-MSH2, six MSH6, and six PMS2) and 13 (3.4%) in 10 non-Lynch syndrome genes (four CHEK2, one each in APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, NBN, PTEN, and RAD51C). Of 21 patients with deleterious mutations in Lynch syndrome genes with tumor testing, 2 (9.5%) had tumor testing results suggestive of sporadic cancer. Of 12 patients with deleterious mutations in MSH6 and PMS2, 10 were diagnosed at age >50 and 8 did not have a family history of Lynch syndrome-associated cancers. Patients with deleterious mutations in non-Lynch syndrome genes were more likely to have serous tumor histology (23.1 vs 6.4%, P=0.02). The three patients with non-Lynch syndrome deleterious mutations and serous histology had mutations in BRCA2, BRIP1, and RAD51C. Current clinical criteria fail to identify a portion of actionable mutations in Lynch syndrome and other hereditary cancer syndromes. Performance characteristics of tumor testing are sufficiently robust to implement universal tumor testing to identify patients with Lynch syndrome. Germline multi-gene panel testing is feasible and informative, leading to the identification of additional actionable mutations.
Similar content being viewed by others


Main
The hallmarks of a hereditary cancer syndrome include early age of diagnosis, multiple affected family members, and an increased lifetime risk of cancers associated with the defined syndrome.1 Lynch syndrome is the prototypical hereditary cancer syndrome in endometrial cancer and accounts for 2–6% of all endometrial cancers.2, 3 Lynch syndrome is caused by autosomal dominant mutations in DNA mismatch repair genes (MLH1, MSH2, MSH6, and PMS2), and patients who carry a germline mutation in one of the mismatch repair genes have a cumulative lifetime risk of endometrial cancer of 20–70%.4, 5, 6, 7 In addition to endometrial cancer, individuals who harbor deleterious mutations in Lynch syndrome genes are at an increased lifetime risk of colorectal, ovarian, gastric, pancreatic, biliary tract, small bowel, and urothelial cancers.8 While less common than Lynch syndrome, Cowden syndrome, characterized by germline mutations in PTEN, also carries an increased lifetime risk of endometrial cancer as high as 28%.9, 10
Hereditary breast and ovarian cancer syndrome is associated with germline mutations in BRCA1 and BRCA2. Patients who harbor germline mutations in BRCA1 and BRCA2 carry a lifetime risk of ovarian cancer (especially high-grade serous carcinoma) of 39–54% and 11–27%, respectively.11, 12, 13 BRCA mutations are also associated with a number of different cancers, most notably breast cancer, pancreatic cancer, and prostate cancer.14 More recently, germline mutations in BRIP1, RAD51D, and RAD51C have been associated with an increased lifetime risk of ovarian cancer ranging from 10–15%,15, 16, 17, 18, 19 with RAD51C and RAD51D associated with the serous subtype. While studies have evaluated the risk of endometrial cancer in hereditary breast and ovarian cancer syndrome, most have focused on serous cancers alone in the Ashkenazi Jewish population and have reported conflicting results.20, 21, 22, 23, 24 Currently, there is no clear evidence that germline mutations in BRCA1, BRCA2, RAD51D, RAD51C, and BRIP1 have a role in hereditary endometrial cancer. Similarly, there is some anecdotal evidence of defects in DNA mismatch repair in breast cancers from women initially suspected as having hereditary breast and ovarian cancer syndrome, but subsequently showing absence of germline mutations in BRCA1 and BRCA2.25 The 25 genes tested in this study were selected based on evidence supporting their role in the development of hereditary cancers (Supplementary Table 1). Although the genes underlying Lynch syndrome are most commonly associated with hereditary endometrial cancer, inclusion of additional hereditary cancer genes allows for the investigation of other genes and the possible association with endometrial cancer.
The traditional approach to identifying the germline mutation associated with a suspected hereditary cancer is to use the patient’s personal and family history to target a specific syndrome and then test for the specific gene or genes associated with that syndrome. Given the overlap of cancer types between syndromes, it is advantageous in certain settings to test for more than one hereditary syndrome simultaneously. Multi-gene panels utilizing next-generation sequencing can be performed rapidly for germline deleterious mutations associated with multiple hereditary cancer syndromes using a single blood sample.26, 27 Evaluation of a multi-gene panel has the potential to provide knowledge regarding the influence of genes other than mismatch repair genes in hereditary endometrial cancer. The objective of the current study was to determine the incidence of germline mutations in Lynch syndrome and other hereditary cancer genes in an unselected cohort of well-characterized endometrial cancer patients. From this study, we can begin to examine the clinical and pathological characteristics of patients with and without germline mutations who have tissue-testing results (immunohistochemistry, microsatellite instability analysis, and MLH1 methylation analysis) suggestive of Lynch syndrome.
Materials and methods
Clinical Data
Following Institutional Review Board approval (PA13-0391), unselected cases of endometrial carcinoma treated at The University of Texas MD Anderson Cancer Center were identified using a departmental tumor bank. Beginning with the most recent cases, endometrial carcinomas were included if the patient was 18 years of age or greater, received treatment at MD Anderson, and had sufficient blood available in the tumor bank for germline analysis. Relevant clinical data were extracted from physician and genetic counselor notes as well as patient intake forms, all available in the electronic medical record. Patients were classified as meeting or not meeting the Society of Gynecologic Oncology 5–10% criteria, which aim to identify patients with a >5–10% chance of having an inherited predisposition to endometrial cancer. These criteria are based on age of endometrial cancer diagnosis, presence of family members with Lynch syndrome-associated cancers, and personal history of Lynch syndrome-associated cancers.28 Tumor testing to screen for potential Lynch Syndrome was performed as previously described in a CLIA-designated clinical laboratory in the Division of Pathology & Laboratory Medicine, MD Anderson Cancer Center.29 Pathological data, immunohistochemistry results for mismatch repair proteins (MLH1, MSH2, MSH6, and PMS2), microsatellite instability (MSI) status, and MLH1 methylation analysis results were abstracted from the pathology report for hysterectomy specimens, if available.
Multi-Gene Next-Generation Sequencing Assay for Germline Assessment
DNA from patient white blood cells was analyzed for germline mutations in a panel of 25 genes associated with hereditary cancer syndromes (MLH1, MSH2, MSH6, PMS2, EPCAM, PTEN, STK11, TP53, BRCA1, BRCA2, APC, ATM, BARD1, BMPR1A, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, bi-allelic MUTYH, NBN, PALB2, RAD51C, RAD51D, and SMAD4) as previously described.26, 27 Sample preparation for next-generation sequencing was performed using the RainDance Thunderstorm emulsion polymerase chain reaction (PCR) system (RainDance Technologies, Billerica, Massachusetts, USA). Next-generation sequencing was performed using the Illumina HiSeq2500 (Illumina, San Diego, California, USA). Large rearrangements were identified using quantitative dosage analysis of the data obtained from next-generation sequencing. In addition, deletions and duplications were identified using a custom microarray comparative genomic hybridization chip (Agilent Technologies, Santa Clara, California, USA). Multiplex ligation-dependent probe amplification analysis for large rearrangements in PMS2 and CHEK2 was performed to distinguish homologous pseudo genes and actual gene regions.30 Variants were classified using American College of Medical Genetics and Genomics recommendations and Myriad Genetics data.31, 32 Gene variants that were deemed deleterious or suspected deleterious were considered mutations. If a patient had a deletion in EPCAM that extended into MSH2, then this patient was counted as one mutation in EPCAM-MSH2. MUTYH variants were considered deleterious only if bi-allelic. Mono-allelic MUTYH mutation carriers were counted and reported separately. Variants of unknown significance were counted and reported separately.
Statistics
The prevalence of deleterious mutations in each of the 25 genes was tabulated and exact 95% confidence intervals were calculated by the Clopper–Pearson method. Demographic, clinical, and pathologic characteristics were compared using the χ2-test for categorical variables and the t-test/ANOVA for continuous variables. As this is primarily a descriptive study, there were no formal adjustments for multiple comparisons. P-values ≤0.05 were considered statistically significant. Analyses were performed using SAS for Windows version 9.3.
Results
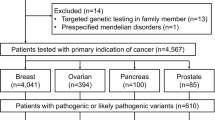
A total of 447 patients with endometrial carcinoma were identified with available clinical data. Sixty-six patients had insufficient DNA for germline testing, resulting in 381 patients included in this analysis. Most (365/381) had previously undergone tumor testing (immunohistochemistry, MSI analysis, and MLH1 methylation) for evaluation of possible Lynch Syndrome. As summarized in Table 1, cases were representative of an endometrial cancer patient population with a mean age of diagnosis 61 years and the majority with stage I or II, grade 1 or 2 endometrioid-type endometrial carcinomas.
The spectrum of germline mutations detected is summarized in Table 2 and Supplementary Tables 2 and 3. Thirty-five patients (9%, 95% confidence interval (CI)=6.48–12.54) had a deleterious mutation in one of the 25 genes examined. Twenty-two patients (6%, 95% CI=3.65–8.61) had a deleterious mutation in Lynch syndrome genes, including 3 MLH1, 5 MSH2, 2 EPCAM-MSH2, 6 MSH6, and 6 PMS2 mutations. Thirteen patients (3%, 95% CI=1.83–5.76) had a deleterious mutation in non-Lynch syndrome genes including four CHEK2 and one each in APC, ATM, BARD1, BRCA1, BRCA2, BRIP1, NBN, PTEN, and RAD51C. Mutations in high-penetrance genes (Lynch syndrome genes, BRCA1, BRCA2, PTEN, and APC) accounted for 74% of the deleterious mutations detected.
Important clinical and pathological characteristics for patients with deleterious germline mutations are summarized in Table 3. We have previously shown that patients with Lynch syndrome germline mutations are more likely to have endometrial tumors centered on the lower uterine segment33 and a lower body mass index.34 Compared with patients with no deleterious mutation, patients with deleterious germline mutations in Lynch syndrome genes were younger at diagnosis (mean 52 vs 62 years, P<0.01), less likely to be overweight (64 vs 86%, P=0.01), more likely to have a tumor in the lower uterine segment (30 vs 8%, P<0.01), and more likely to meet the Society of Gynecologic Oncology guidelines for genetic assessment referral (59 vs 24%, P=0.01). Of note, three patients (14%) with deleterious germline mutations in Lynch syndrome genes were diagnosed at age >60 years, and 41% of the patients with a Lynch syndrome deleterious mutation did not meet the Society of Gynecologic Oncology criteria for genetic assessment referral. Twenty-one of 22 patients with deleterious mutations in Lynch syndrome genes had available tumor-testing results. Two (10%) of these patients had a deleterious mutation (one PMS2 and one MSH6) and intact immunohistochemistry expression of mismatch repair proteins without MSI, suggestive of a sporadic cancer. Patients with deleterious germline mutations in non-Lynch syndrome genes were more likely to have endometrial carcinomas with serous histology (23 vs 6%, P=0.02) than patients without a deleterious mutation. No other predictors of deleterious mutations in non-Lynch syndrome genes were identified.
Table 4 summarizes age at diagnosis, tumor histology, and family history for patients with deleterious germline mutation in non-Lynch syndrome genes. Of the 13 patients with deleterious mutations in non-Lynch syndrome genes, 5 (39%) were of non-endometrioid histology. Neither of the deleterious mutations in BRCA1 or BRCA2 was clinically recognized prior to this study. Both patients with a deleterious mutation in PTEN and APC previously presented with clinical histories consistent with Cowden syndrome and familial adenomatous polyposis, respectively. The patient with a deleterious mutation in PTEN presented with a personal history of uterine and breast cancer and autism, as well as a family history of breast, colon, and thyroid cancer. The patient with a deleterious mutation in APC had a screening colonoscopy following her uterine cancer diagnosis given a family history of colon cancer. On screening colonoscopy, the patient was found to have polyposis consistent with familial adenomatous polyposis. Note that the ages of endometrial cancer diagnosis (35 and 28 years, respectively) for these two women were substantially younger than the mean age for women with a deleterious mutation in a Lynch syndrome gene (52 years).
Tumor testing (immunohistochemistry for DNA mismatch repair proteins, MSI analysis, and MLH1 methylation analysis) has emerged as a useful tissue screening tool to help identify patients at risk for harboring a Lynch syndrome mutation. It is well known that patients who are tissue screen-positive may not subsequently have a germline Lynch syndrome gene mutation detected. Of the 365 patients that had tumor testing performed, 51 (14%) were tissue screen-positive defined as loss of MLH1 expression (without MLH1 methylation) or loss of MSH2, MSH6, or PMS2 by immunohistochemistry. Patients with MSI-high tumors lacking MLH1 methylation but with intact immunohistochemistry expression of mismatch repair proteins were also considered tissue screen-positive. Thirty of these patients (63%) had tumor testing results suggestive of Lynch syndrome, but were found to have no germline mutation in Lynch syndrome genes. As shown in Table 5, patients with deleterious germline mutations in Lynch syndrome genes were diagnosed at a younger age compared with tissue screen-positive but Lynch syndrome germline-negative patients (mean 52 vs 60 years, P<0.01). However, there were no significant differences in BMI, tumor location, and family history between patients with deleterious mutations in Lynch syndrome genes, and screen-positive patients with no identifiable deleterious mutation. Interestingly, when examining BMI, tumor location (lower uterine segment vs corpus), the Society of Gynecologic Oncology criteria, and family history of Lynch-associated cancer, there is a graded difference between the Lynch syndrome deleterious mutations, tissue screen-positive/germline Lynch syndrome deleterious mutation-negative, and sporadic cancer groups. For example, the percentage of screen-positive/germline Lynch syndrome deleterious mutation-negative patients with a family history of Lynch-associated cancer was 42%, greater than that for the sporadic patients (33%) but less than that for the Lynch syndrome deleterious mutation patients (59%). In addition, the endometrial cancer patients who are tissue screen-positive but Lynch syndrome germline mutation-negative tend to have clinical and pathological features which are intermediate between patients with identified Lynch syndrome germline mutations and patients with sporadic endometrial cancer (no Lynch syndrome mutation and not tissue screen-positive).
Table 6 summarizes age of diagnosis and family history for the individual Lynch syndrome mutations. For deleterious mutations in MLH1, MSH2, and EPCAM-MSH2, 80% of patients were diagnosed at age <50 years, 90% of patients met the Society of Gynecologic Oncology criteria for genetic assessment referral, and 90% have a documented family history of Lynch syndrome-associated cancer. Conversely, for deleterious mutations in MSH6 and PMS2, 83% of patients were diagnosed at age >50 years, 67% did not meet the Society of Gynecologic Oncology criteria, and 67% did not have a documented family history of Lynch syndrome-associated cancers.
The sensitivity and specificity of the Society of Gynecologic Oncology criteria for identifying patients with deleterious mutation in Lynch syndrome genes were 59 and 75%, respectively, compared with 90 and 91% for tumor testing with immunohistochemistry for mismatch repair proteins, MSI analysis, and MLH1 methylation for cases involving MLH1 protein loss by immunohistochemistry. As expected, tumor testing with immunohistochemistry, MSI, and MLH1 methylation, did not identify patients with non-Lynch syndrome gene mutations.
Discussion
To our knowledge, this is the first report of multi-gene hereditary cancer panel testing in an unselected endometrial carcinoma cohort. The hereditary cancer panel incorporated genes known to be associated with hereditary endometrial cancer in addition to other known hereditary cancer syndromes. Similar to previous studies, we found that 6% of this population has a deleterious germline mutation consistent with Lynch syndrome. Thirteen patients were found to have a deleterious mutation in non-Lynch syndrome genes. Interestingly, 5 of these 13 patients had endometrial carcinomas that were non-endometrioid histology; these are clinically the most aggressive endometrial cancers with the highest risk of mortality. Although there is a well-established association between BRCA mutations and serous ovarian carcinomas, previous studies that have evaluated the role of BRCA mutations in uterine serous carcinomas have produced conflicting results. Three studies have reported on the incidence of BRCA founder mutations in patients with uterine serous carcinoma and Ashkenazi Jewish ancestry. Biron-Shental et al23 evaluated 22 patients with uterine serous cancer and found that 6 patients had a germline mutation in BRCA1 or BRCA2, accounting for 27% of patients. Lavie et al22 also reported eight BRCA1 and BRCA2 mutations in a series of 59 uterine serous cancers. Differently, Levine et al20 evaluated a series of 199 Ashkenazi Jewish patients with endometrial carcinoma that included 17 serous carcinomas. There were three BRCA germline mutations identified overall in this population, but no mutations were identified in the serous cancers specifically.20 Of note, one of the BRCA mutations identified in our study was a founder mutation, but neither reported Ashkenazi Jewish ancestry. Two additional studies have evaluated BRCA mutations in the general uterine serous cancer patient population. Goshen et al24 reported no germline mutations in BRCA1 or BRCA2 in 56 patients with serous uterine cancer. Pennington et al21 evaluated a larger series of 151 uterine serous cancers for germline mutations in BRCA1, BRCA2, MLH1, MSH2, MSH6, PMS2, and 10 Fanconi anemia-BRCA pathway genes. In their cohort, 4.7% of patients were found to have germline mutations, with three BRCA1 mutations, two CHEK2 mutations, and no BRCA2 mutations identified.21 Although Pennington et al21 included BRIP1 and RAD51C in their analysis, there were no mutations identified in their patient population. There were 26 serous cancers included in our patient cohort and 3 had a germline mutation in genes associated with hereditary ovarian cancer (BRCA2, RAD51C, and BRIP1), accounting for 11.5% of this patient population.13, 16, 17 The patient with a BRCA2 mutation reported one family member with a postmenopausal breast cancer and both patients with a RAD51C and BRIP1 mutations reported a family history of pancreatic cancer. None of these patients reported a family history of ovarian cancer and were not previously identified clinically for genetic assessment referral. One patient with BRCA1 germline mutation had an endometrioid-type endometrial carcinoma; she also reported no family history of cancer.
A recent report by Yurgelun et al27 evaluated the role of multi-gene panel testing in patients with a clinical history consistent with Lynch syndrome. The majority of patients included in this large registry-based series presented with colorectal cancer, but there were 292 patients with endometrial cancer included. Of these, two BRCA1, two BRCA2, one CHEK2, and one ATM germline mutations were identified.27 This study differs from the current patient cohort in that the patient population had higher risk for germline mutations given that all patients included had a clinical history suggestive of Lynch syndrome. In addition, given that this was a registry-based study, there was no information available regarding the histologies of the individual mutation carriers. Despite these differences, this study reinforces the findings that a proportion of endometrial cancer patients have a germline mutation in BRCA1, BRCA2, and other hereditary cancer predisposition genes. Larger studies, in uterine serous cancers specifically, are needed to address the role of hereditary breast and ovarian cancer genes in hereditary endometrial cancer. It is currently unclear if female relatives of uterine serous carcinoma patients with germline mutations in BRCA1, BRCA2, or other genes are also at risk of developing this aggressive endometrial malignancy. Based on the results of our study and the cumulative published literature, testing patients with serous uterine cancer for germline mutations in multiple hereditary cancer genes has potential clinical implications that include interventions for breast or ovarian cancer.
Four patients (endometrioid histology × 3; clear cell histology × 1) were found to have a mutation in CHEK2, making it the most common non-Lynch syndrome deleterious mutation identified in this patient cohort. A germline mutation in CHEK2 (1100delC) was first identified in 2002 and since then has been associated with an increased risk of breast cancer.35 In addition, CHEK2 mutations have also been associated with prostate, thyroid, renal, and questionably colorectal cancer.36, 37, 38 Pennington et al21 did identify two CHEK2 mutations in their series of patients with uterine serous carcinoma, but no studies to date have found CHEK2 mutations associated with other endometrial cancer histologies.21 Two of four patients with CHEK2 mutations in our study reported a family history of breast cancer. The risk of breast cancer associated with CHEK2 has been shown to be higher in individuals with a family history of breast cancer. NCCN guidelines recommend annual breast MRI screening for patients who have a lifetime risk of 20% or greater based on gene mutation status or family cancer history. In our study population, eight patients with deleterious germline mutation in BRCA1, BRCA2, CHEK2, ATM, and PTEN would fall under this recommendation highlighting the clinical implications of finding germline mutations outside of Lynch syndrome in this patient population.39
One strength of our study is the ability to compare germline mutation analysis with tumor-testing results to differentiate germline Lynch syndrome-positive cases from screen-positive patients with no identifiable Lynch syndrome germline mutation or so-called 'Lynch-like syndrome (LLS)' cases. In our study, the only significant difference between germline-positive cases and screen-positive cases was mean age of diagnosis, 51.7 vs 59.6, respectively. There were no differences in BMI, tumor location, whether patients met the Society of Gynecologic Oncology criteria or documented family history of Lynch syndrome-associated cancers when comparing cases with Lynch syndrome deleterious mutation and screen-positive patients. This raises an important clinical question of how to best manage patients with endometrial cancer that are tissue screen-positive, germline Lynch syndrome mutation-negative. Recommending more frequent colonoscopy than the general population is an important option.
There have been recent advances that help explain some of these cases, including cases with a loss of expression of mismatch repair protein, but no germline mutation identified. Rhees et al40 reported a novel inversion of exons 1–7 of MSH2 in 6/10 cases in which immunohistochemistry showed loss of MSH2 protein expression with no identifiable deleterious mutation by germline testing. While 5 patients in the current study had MSH2 mutations, an additional 12 patients had loss of MSH2 expression on immunohistochemistry with no identifiable mutation in MSH2. MSH2 inversion analysis was not incorporated into the current gene panel and may explain a portion of these cases. In addition, two groups have reported mismatch repair gene somatic mutations in carcinomas with loss of mismatch repair proteins on immunohistochemistry but no germline mutation identified.41, 42 Based on these findings, MSH2 inversion testing and somatic mutation screening could potentially provide an explanation for a proportion of tissue screen-positive, germline-negative cases. A stepwise approach incorporating these techniques may help to prevent unnecessary increased colorectal screening, as well as ease patient anxiety for those who have no identifiable germline mutation via routine germline testing.
In our study, of 22 patients found to have Lynch syndrome germline mutations, 55% had MSH6 and PMS2 mutations. Previous studies have shown that women who carry MSH6 mutations have an increased risk of endometrial cancer compared with those with MLH1 and MSH2 mutations.43, 44 More importantly, patients with MSH6 and PMS2 mutations did not present with clinical histories that are classically seen with Lynch syndrome. In contrast to patients with MLH1, MSH2, and EPCAM-MSH2 mutations, the majority of patients with MSH6 and PMS2 mutations were diagnosed at age >50 years, did not meet the Society of Gynecologic Oncology criteria, and did not have a documented family history of Lynch syndrome-associated cancers. This raises the question of whether there are two distinct presentations of Lynch syndrome-associated endometrial cancer. So-called 'typical Lynch syndrome-associated endometrial cancer' presents with early age of endometrial cancer diagnosis, significant family history for Lynch syndrome-associated cancers, and multiple cancers within the same patient. These patients are more likely to have mutations in MLH1 and MSH2 according to our data. In contrast, women with 'atypical Lynch syndrome-associated endometrial cancer' present at later age of diagnosis and lack significant family history. According to our data, as many as 67% of endometrial cancer patients with MSH6 or PMS2 germline mutations would not be identified using Society of Gynecologic Oncology clinical screening criteria. One approach that has been proposed is universal tissue screening for all newly diagnosed endometrial cancer cases. Even this approach does not capture all patients with germline Lynch syndrome mutations and is not expected to identify patients with mutation in hereditary cancer genes outside of Lynch syndrome. In our study, there were two Lynch syndrome germline mutations identified, one PMS2, and one MSH6, associated with intact positive immunohistochemistry expression of mismatch repair proteins. There was insufficient tumor for MSI analysis for these two patients.
One limitation of our study is that clinical characteristics were collected retrospectively from patient questionnaires and dictated physician notes in the electronic medical record, which could lead to incomplete personal and family histories. In addition, all patients were collected from a large referral cancer center, which may have an overall higher risk of hereditary cancer than the general endometrial cancer patient population. With a total of 35 germline mutations identified, making definitive conclusions regarding individual genes is difficult. Our data do suggest that a patient population more enriched in non-endometrioid endometrial carcinomas may be enriched in germline mutations, particularly in non-Lynch syndrome genes.
Our conclusions from this study are three-fold. First, a significant percentage of endometrial cancer patients with Lynch syndrome are not recognized using current clinical criteria, as they present with later age of diagnosis or with limited or no family history of Lynch syndrome-associated cancers. Second, the performance characteristics of tumor testing are sufficiently robust to utilize universal tumor testing in all newly diagnosed endometrial carcinomas to identify patients with Lynch syndrome who may be missed by current referral guidelines. Tumor testing does not identify mutation carriers beyond Lynch syndrome. Third, germline multi-gene hereditary cancer panel testing is feasible and informative in a large series of unselected endometrial carcinoma cases. In addition to the Lynch mutation carriers, 2% of patients were identified with cancer predisposition gene mutation with available NCCN management guidelines. The clinical relevance of identifying these additional gene mutation carriers should be explored in future studies.
References
Garber JE, Offit K . Hereditary cancer predisposition syndromes. J Clin Oncol 2005;23:276–292.
Leenen CHM, van Lier MGF, van Doorn HC et al. Prospective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer ≤70 years. Gynecol Oncol 2012;125:414–420.
Backes FJ, Cohn DE . Lynch syndrome. Clin Obstet Gynecol 2011;54:199–214.
Dunlop MG, Farrington SM, Carothers AD et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Gen 1997;6:105–110.
Aarnio M, Sankila R, Pukkala E et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999;81:214–218.
Stoffel E, Mukherjee B, Raymond VM et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterol 2009;137:1621–1627.
Baglietto L, Lindor NM, Dowty JG et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010;102:193–201.
Vasen HFA, Blanco I, Aktan-Collan K et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;62:812–823.
Pilarski R, Burt R, Kohlman W et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 2013;105:1607–1616.
Bubien V, Bonnet F, Brouste V et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 2013;50:255–263.
Ford D, Easton DF, Stratton M et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 1998;62:676–689.
Antoniou A, Pharoah PDP, Narod S et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003;72:1117–1130.
King M-C, Marks JH, Mandell JB et al. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003;302:643–646.
Levy-Lahad E, Friedman E . Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer 2007;96:11–15.
Loveday C, Turnbull C, Ramsay E et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 2011;43:879–882.
Loveday C, Turnbull C, Ruark E et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat Genet 2012;44:475–476.
Rafnar T, Gudbjartsson DF, Sulem P et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet 2011;43:1104–1107.
Walsh T, Casadei S, Lee MK et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 2011;108:18032–18037.
Song H, Dicks E, Ramus SJ et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol 2015;33:2901–2907.
Levine DA, Lin O, Barakat RR et al. Risk of endometrial carcinoma associated with BRCA mutation. Gynecol Oncol 2001;80:395–398.
Pennington KP, Walsh T, Lee M et al. BRCA1, TP53, and CHEK2 Germline mutations in uterine serous carcinoma. Cancer 2013;119:332–338.
Lavie O, Ben-Arie A, Segev Y et al. BRCA Germline mutations in women with uterine serous carcinoma-still a debate. Int J Gynecol Cancer 2010;20:1531–1534.
Biron-Shental T, Drucker L, Altaras M et al. High incidence of BRCA1–2 germline mutations, previous breast cancer and familial cancer history in Jewish patients with uterine serous papillary carcinoma. Eur J Surg Oncol 2006;32:1097–1100.
Goshen R, Chu W, Elit L et al. Is uterine papillary serous adenocarcinoma a manifestation of the hereditary breast–ovarian cancer syndrome? Gynecol Oncol 2000;79:477–481.
Buerki N, Gautier L, Kovac M et al. Evidence for breast cancer as an integral part of Lynch syndrome. Genes Chromosomes Cancer 2012;51:83–91.
Tung N, Battelli C, Allen B et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015;121:25–33.
Yurgelun MB, Allen B, Kaldate RR et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 2015;149:604–613.
Lancaster JM, Powell CB, Kauff ND et al. Society of Gynecologic Oncologists education committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol 2007;107:159–162.
Bruegl AS, Djordjevic B, Batte B et al. Evaluation of clinical criteria for the identification of Lynch syndrome among unselected patients with endometrial cancer. Cancer Prev Res 2014;7:686–697.
Judkins T, Leclair B, Bowles K et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer 2015;15:215.
Richards CS, Bale S, Bellissimo DB et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 2008;10:294–300.
Eggington JM, Bowles KR, Moyes K et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet 2014;86:229–237.
Westin SN, Lacour RA, Urbauer DL et al. Carcinoma of the lower uterine segment: a newly described association with Lynch syndrome. J Clin Oncol 2008;26:5965–5971.
Lu KH, Schorge JO, Rodabaugh KJ et al. Prospective determination of prevalence of Lynch syndrome in young women with endometrial cancer. J Clin Oncol 2007;25:5158–5164.
Meijers-Heijboer H, van den Ouweland A, Klijn J et al. Low-penetrance susceptibility to breast cancer due to CHEK2[ast]1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002;31:55–59.
Dong X, Wang L, Taniguchi K et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet 2003;72:270–280.
Cybulski C, Górski B, Huzarski T et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004;75:1131–1135.
Meijers-Heijboer H, Wasielewski M, Wagner A et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am J Hum Genet 2003;72:1308–1314.
Daly M, Pilarski R, Axilbund JE et al. Genetic/Familial High-Risk Assessment: Breast and Ovarian. Version 2.2015. NCCN Clinical Practice Guidelines in Oncology 2015; http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf.
Rhees J, Arnold M, Boland CR . Inversion of exons 1–7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam Cancer 2014;13:219–225.
Haraldsdottir S, Hampel H, Tomsic J et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterol 2014;147:1308–1316 e1.
Geurts-Giele WRR, Leenen CHM, Dubbink HJ et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol 2014;234:548–559.
Baglietto L, Lindor NM, Dowty JG et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010;102:193–201.
Hendriks YMC, Wagner A, Morreau H et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterol 2004;127:17–25.
Acknowledgements
This study was supported by NIH Research Training Grant (KLR and ASB) T32 CA101642; NIH SPORE in Uterine Cancer (RRB) NIH 2P50 CA098258.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
BAA, EPE, NS, and AR Hartman were employed by Myriad Genetics, Inc. at the time of this study and received salaries and stock options as compensation. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Ring, K., Bruegl, A., Allen, B. et al. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol 29, 1381–1389 (2016). https://doi.org/10.1038/modpathol.2016.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2016.135
This article is cited by
-
Endometrial cancer
Nature Reviews Disease Primers (2021)
-
Stakeholders’ views of integrating universal tumour screening and genetic testing for colorectal and endometrial cancer into routine oncology
European Journal of Human Genetics (2021)
-
In search for biomarkers and potential drug targets for uterine serous endometrial cancer
Journal of Cancer Research and Clinical Oncology (2021)
-
Prevalence of mutations in BRCA and MMR genes in patients affected with hereditary endometrial cancer
Medical Oncology (2021)
-
Modern day screening for Lynch syndrome in endometrial cancer: the KEM experience
Archives of Gynecology and Obstetrics (2021)
