
Abstract
Cutaneous neuroendocrine (Merkel cell) carcinoma most often arises de novo in the background of a clonally integrated virus, the Merkel cell polyomavirus, and is notable for positive expression of retinoblastoma 1 (RB1) protein and low expression of p53 compared with the rare Merkel cell polyomavirus-negative Merkel cell carcinomas. Combined squamous and Merkel cell tumors are consistently negative for Merkel cell polyomavirus. Little is known about their immunophenotypic or molecular profile. Herein, we studied 10 combined cutaneous squamous cell and neuroendocrine carcinomas for immunohistochemical expression of p53, retinoblastoma 1 protein, neurofilament, p63, and cytokeratin 20 (CK20). We compared mutation profiles of five combined Merkel cell carcinomas and seven ‘pure’ Merkel cell carcinomas using targeted next-generation sequencing. Combined tumors were from the head, trunk, and leg of Caucasian males and one female aged 52–89. All cases were highly p53- and p63-positive and neurofilament-negative in the squamous component, whereas RB1-negative in both components. Eight out of 10 were p53-positive, 3/10 p63-positive, and 3/10 focally neurofilament-positive in the neuroendocrine component. Six out of 10 were CK20-positive in any part. By next-generation sequencing, combined tumors were highly mutated, with an average of 48 mutations per megabase compared with pure tumors, which showed 1.25 mutations per megabase. RB1 and p53 mutations were identified in all five combined tumors. Combined tumors represent an immunophenotypically and genetically distinct variant of primary cutaneous neuroendocrine carcinomas, notable for a highly mutated genetic profile, significant p53 expression and/or mutation, absent RB1 expression in the context of increased RB1 mutation, and minimal neurofilament expression.
Similar content being viewed by others

Main
Distinct oncogenetic differences between Merkel cell polyomavirus-related cutaneous neuroendocrine (Merkel cell) carcinoma and Merkel cell polyomavirus-negative Merkel cell carcinoma have recently been established,1, 2, 3, 4, 5 suggesting heterogeneity among primary cutaneous neuroendocrine carcinomas.
Merkel cell carcinoma, the eponym for primary cutaneous neuroendocrine carcinoma, is generally regarded as a uniform clinical/histogenetic entity, in which variation of histopathology has had little impact on prognosis or treatment.6, 7, 8 The vast majority of Merkel cell carcinoma express epithelial antigens, such as cytokeratins, (most characteristically cytokeratin 20 (CK20)), neuroendocrine markers (chromogranin, synaptophysin, CD56), and neurofilament.6, 7, 9, 10, 11 The identification of a clonally integrated polyomavirus, the Merkel cell polyomavirus, present in the majority of Merkel cell carcinoma, but absent in a subset, has set the stage for a re-evaluation of differences among histopathologic variants of Merkel cell carcinoma.
Merkel cell polyomavirus has repeatedly been identified in 55–90% of Merkel cell carcinoma by both protein immunohistochemistry using an antibody (clone CM2B4) to the viral large T antigen, and by PCR.3, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 More recently, it has been determined that Merkel cell polyomavirus-positive cases show strong retinoblastoma 1 (RB1) immunohistochemical labeling but rare p53 expression, whereas negative cases show weak RB1 expression, but more frequent p53-protein expression.4, 24 Recent studies have suggested a possible survival difference between these two subsets of disease.4, 25, 26, 27 Although histologically combined lesions of Merkel cell carcinoma with non-Merkel cell carcinoma tumors, in particular, squamous cell carcinoma, have been historically accepted as variants of Merkel cell carcinoma,8 it remains to be elucidated, if such tumors might derive from a distinct histogenetic precursor cell, or if they might have unique prognostic and therapeutically exploitable characteristics. Given the absence of Merkel cell polyomavirus in reported series of combined squamous cell carcinoma/Merkel cell carcinoma,1, 2, 13, 17, 19 it seemed likely that we would find more differences to distinguish these combined tumors from pure Merkel cell carcinoma. Furthermore, the identification of strong p53 and p63 labeling in a case of a combined squamous cell carcinoma in situ/Merkel cell carcinoma in situ, as well as unexpectedly absent neurofilament labeling,28 supported this hypothesis.
To identify the distinguishing characteristics of combined squamous cell carcinoma/Merkel cell carcinoma, we examined the immunohistochemical expression of CK20, p53, neurofilament, and RB1 in these tumors. In the context of reports of p63 expression correlating with outcome29, 30, 31, 32 we also assessed p63 in this series of combined tumors. Targeted next-generation sequencing was performed, assessing a panel of 230 tumor-associated genes with select combined tumors and an equal number of ‘pure’ tumors as controls. Clinical data were reviewed for unique characteristics associated with tumor subsets.
Materials and methods
Eleven patients’ slides and corresponding blocks with combined squamous cell carcinoma/Merkel cell carcinoma were identified and retrieved from the pathology archive of Memorial Sloan-Kettering Cancer Center, in addition to one case with squamous cell carcinoma in situ/Merkel cell carcinoma in situ previously reported.28 Diagnoses were confirmed by two dermatopathologists (MPP & KJB). Charts were reviewed for demographics, course of disease, and other significant medical history.
Histologic examination of 15 specimens (5 biopsies and 10 excisions) from 10 patients who had tissue available for immunohistochemical study, was performed on formalin-fixed paraffin-embedded tissue sections stained with hematoxylin & eosin using standard protocols. Morphologic features were examined, including pattern and relationship of Merkel cell carcinoma and squamous cell carcinoma tumor components, presence or absence of invasive tumor, and cellular morphology.
Immunohistochemical stains were performed using antibodies to the Merkel cell polyomavirus large T antigen (CM2B4), CK20, neurofilament, p53, p63, and RB1 proteins (Table 1), with appropriate positive and negative controls, on specimens available from 10 of the patients. Immunohistochemical stains were reviewed independently by two dermatopathologists. The pattern of staining of CK20 and neurofilament was noted (perinuclear dot-like, membranous or diffuse). For p53, p63 and RB1, nuclear labeling was accepted as positive, and a tumor component was considered positive if at least 75% of cells exhibited intense staining. For CK20 and neurofilament, a fully positive stain required at least 50% of tumor cells labeling. Focal staining was noted separately, if present.
DNA Extraction
Seven specimens from seven patients with combined squamous cell carcinoma/Merkel cell carcinoma were selected, each of which appeared to have abundant tumor tissue on review of H&E-stained sections, and which were available for genomic analysis (one additional specimen/patient not studied for immunohistochemical features was included; only tissue curls were available for molecular analysis). Seven additional specimens from seven patients with pure Merkel cell carcinoma, 6/7 of which were confirmed to be positive for Merkel cell polyomavirus by immunohistochemical analysis, were selected. For tissue specimens, DNA was extracted and purified according to the protocol for DNA isolation from formalin-fixed paraffin-embedded tissue, from Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis (Agilent technologies, Santa Clara, CA, USA). Genomic DNA was extracted from peripheral blood samples using the Blood and body fluid spin protocol with QIAamp DNA blood mini kit according to the manufacturer’s protocol (Qiagen, Hilden, Germany). Control ‘normal’ genomic tissue was available for all 14 cases, from prior formalin-fixed paraffin-embedded surgical material. One patient’s combined tumor tissue yielded inadequate DNA for analysis. A second patient’s combined tumor yielded no mutations or copy number alterations; on re-review of slides it was found that the tumor tissue was exhausted prior to DNA extraction.
Targeted Next-Generation Sequencing
We profiled genomic alterations in 230 key cancer-associated genes using our IMPACT assay (Integrated Mutation Profiling of Actionable Cancer Targets), using custom oligonucleotides for hybridization capture of all protein-coding exons and select introns of these genes. Bar-coded sequence libraries were prepared (New England Biolabs, Kapa Biosystems) and exon capture was performed (Nimblegen SeqCap) as previously described.33 In total, 60–100 ng of genomic DNA was input for library construction. Libraries were pooled at equimolar concentrations (100 ng per library) and input to a single-exon capture reaction. To prevent off-target hybridization, we spiked in a pool of blocker oligonucleotides complementary to the full sequences of all bar-coded adaptors to a final total concentration of 10 micromolar. DNA was subsequently sequenced on an Illumina HiSeq 2500 to generate paired-end 75-bp reads. Sequence data were demultiplexed using CASAVA, and reads were aligned to the reference human genome (hg19) using the Burrows-Wheeler Alignment tool.34 Local realignment and quality score recalibration were performed using the Genome Analysis Toolkit (GATK) according to GATK best practices.35 We achieved a mean unique sequence coverage of 288X per sample. Single-nucleotide variants were called using MuTect36 and retained if the variant allele frequency in the tumor was more than five times that in the matched normal. Indels were called using the SomaticIndelDetector tool in GATK. All candidate mutations and indels were reviewed manually using the Integrative Genomics Viewer.37
Results
Our data support the hypothesis that the mutational pattern and protein expression profile of combined squamous cell carcinoma/Merkel cell carcinoma is distinct from Merkel cell polyomavirus-positive tumors (Table 3), and argues that they arise from a distinct histogenetic precursor likely via an Merkel cell polyomavirus-independent molecular pathway.
Clinical
Clinical features of patients with tumors examined by immunohistochemistry are shown in Table 2. Of specimens submitted for genomic sequencing, five specimens of squamous cell carcinoma/Merkel cell carcinoma were successful in providing analyzable data. These five patients were all Caucasian, 5/5 male, aged 52–79 (median 66). All five tumors examined were primary tumors from the head (3), thigh (1), or chest (1). Sites of primary tumors from pure controls included ear (1), arm (3), buttock (2), and an axillary tumor of unknown primary (1). Substrate for molecular analysis of the pure tumors included one arm lesion, one ear lesion, two buttock lesions, and three lymph node tumors.
Histopathology
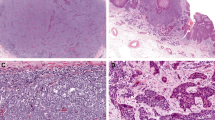
Histopathologic review showed variability in the composition of the squamous and neuroendocrine components. In six cases both components were invasive (Figure 1a–d); in one case both components were in situ,28 and in three cases the squamous cell carcinoma was in situ, whereas the neuroendocrine component involved the dermis. In nine cases, the neuroendocrine component showed a medium-to-large cell morphology (Figure 1a, c and d); the tenth case combined small-cell cytology including nuclear molding and crush artifact and well-differentiated invasive squamous cell carcinoma (Figure 1b).

(a) Case no.1. Hematoxylin & eosin. Superficial squamous cell carcinoma with focal transition to neuroendocrine carcinoma, within basal layers of the epidermis, and within the dermis of tangientially sectioned dermal papillae. (b) Case no. 2. Hematoxylin & eosin stain showing well-differentiated islands of squamous cell carcinoma abutting small islands and cords of neuroendocrine tumor. (c) Case no.3. Hematoxylin & eosin stain. Poorly differentiated small round blue cell tumor with squamous islands. (d) Case no. 5. Hematoxylin & eosin stain shows the small cell tumor transitioning into keratinizing squamous eddies.
Immunohistochemistry
Immunohistochemical findings are presented in Table 3. All cases tested were negative for mAb CM2B4 to Merkel cell polyomavirus large T antigen. CK20 was positive in 6/10 cases in one or both components, and negative in 4/10 cases (Figure 2a). In one case CK20 was positive in the purported primary tumor, but negative in the metastasis. Neurofilament was negative in all 10 cases in the squamous cell carcinoma component (100%), and in 5/10 cases in the neuroendocrine component (Figure 2b). Although 3/10 cases showed moderate dot-like neurofilament labeling of the neuroendocrine area, two were focally and weakly positive. All cases were strongly and diffusely positive for p53 in the squamous component (10/10, 100%). Only 2/10 cases (20%) were p53-negative in the neuroendocrine component. All cases were RB1-negative (0/10, 100%) in the neuroendocrine component (Figure 2d). One showed RB1 staining in the squamous cell component. All 10 cases (100%) were positive for p63 in the squamous component (whether squamous cell carcinoma in situ or squamous cell carcinoma), however, only 3/10 cases (30%) were positive for p63 in the neuroendocrine component. In one case, an area of poorly differentiated squamous cell carcinoma showed focal absence of p63, correlating with absent p63 in the neuroendocrine component.

(a) Case no. 6. Immunohistochemical stain for cytokeratin 20 is negative within neuroendocrine carcinoma. (b) Case no. 6. Immunohistochemical stain for neurofilament is negative within the neuroendocrine carcinoma. (c) Case no. 6. P53 immunohistochemical stain strongly labels both squamous cell and neuroendocrine tumor components. (d) Case no. 6. Immunohistochemical stain for retinoblastoma 1 showing near complete negativity in the neuroendocrine carcinoma. Rare small lymphocytes are positive.
Molecular
Five combined tumors and seven pure tumors provided sufficient DNA for analysis in tumor and control tissue.
Analysis of the two sets of tumors showed that the average number of mutations (single-nucleotide variants, dinucleotide variants, and indels) per sample was 38 times higher for combined tumors vs pure tumors (mean 32 vs 0.85, median 29 vs 1) (Figure 3). The mean number of mutations per megabase was 48 for combined tumors vs 1.25 for pure tumors. Single-nucleotide variants alone averaged 39 per megabase for combined tumors, vs 1 per megabase in pure tumors. Overall, C–T substitutions comprised 78% (103/132) of single-nucleotide variants in the combined tumors, consistent with an ultraviolet (UV)-light mutation signature, vs 40% (2/5) of mutations in pure tumors. RB1 and p53 were the most commonly involved genes to harbor mutations in combined tumors (5/5), whereas none of the pure tumors had RB1 or p53 mutations. Seven different mutations in RB1 were noted (all unique alterations). Eight mutations in p53 were noted, one of which was recurrent in two samples. Five of these were C–T substitutions, suggestive of a UV phenotype. Three cases each showed two different RB1 and/or p53 mutations. NOTCH1 and ERBB4 genes were mutated in 4/5 combined tumors but no pure tumors.

Total number of tumor mutations in combined vs pure Merkel cell carcinoma. Tumor numbers 1–5 represent combined squamous cell and neuroendocrine carcinoma. 6–12 represent pure Merkel cell carcinoma.
Discussion
Our findings show that combined squamous cell and neuroendocrine carcinoma differ from pure Merkel cell carcinoma by protein expression as well as genetically, not just by morphology and the absence of Merkel cell polyomavirus. We identified significant differences in predicted positive vs negative expression of p53 and RB1 protein, and neurofilament, particularly when the squamous component was evaluated (Tables 1 and 3). We also identified differences in CK20 expression. On the molecular level, we demonstrated a highly mutated genetic profile for all combined tumors compared with notably ‘quiet’ pure Merkel cell carcinoma. Furthermore, only the combined tumors showed mutations in p53 and RB1 genes consistent with a UV-related pathogenesis, as well as frequently expressed non-recurrent mutations in select other genes, eg NOTCH1, suggestive of mutational hotspots in these tumors but not in controls.
Since the discovery of the clonal integration of Merkel cell polyomavirus in Merkel cell carcinoma in 2008,15 it has become apparent that ~20% of Merkel cell carcinoma show no evidence of associated Merkel cell polyomavirus, neither by PCR nor by immunohistochemistry. In our earlier series we found 88% PCR positivity for Merkel cell polyomavirus and positive immunohistochemistry for the viral large T antigen CM2B4 in 67%. Within the group of Merkel cell carcinoma negative for antibody CM2B4, we found that squamous cell carcinoma/Merkel cell carcinoma showed complete negativity for Merkel cell polyomavirus by PCR and immunohistochemistry.13 Others have confirmed these findings.1, 2, 17, 19
Sihto et al showed that 11/23 (48%) Merkel cell polyomavirus-negative Merkel cell carcinoma carried p53 mutations in contrast to 0/16 Merkel cell polyomavirus-positive tumors. Furthermore, mAb CM2B4+ Merkel cell carcinoma expressed RB1, whereas mAb CM2B4 tumors, for the most part, did not.4 Waltari et al reported that of eighteen p53-protein positive Merkel cell carcinomas, 50% harbored no viral DNA, vs 18% of p53-protein negative Merkel cell carcinoma, and that Merkel cell polyomavirus DNA copy number decreased as p53-positive cells increased. Moreover, p53 expression was associated with poor disease-specific survival.5 Bhatia et al demonstrated that viral abundance correlated directly with RB1 protein, and was associated with less p53 expression as well as longer survival.3
Historically the convention has been to consider histopathologically biphenotypic cutaneous neuroendocrine tumors as variations of Merkel cell carcinoma. This is predicated on a report by Tang and Toker in which a tumor with areas of unequivocal squamous cell carcinoma was discussed;38 in the same report, the authors rejected epidermis, sweat gland ducts and hair follicles as a possible tissue of origin for Merkel cell carcinoma. In the following five years, at least 22 similar cases were reported.39, 40, 41 Tang and Toker eventually concluded that ‘the spectrum of malignant Merkel cell neoplasm is expanding’.41 Such cases have subsequently been reported in both small and large-scale analyses, exemplifying the morphologic diversity of this tumor.6, 8, 42, 43, 44, 45, 46, 47, 48, 49
In the last 30 years, squamous cell carcinoma/Merkel cell carcinoma has been treated as a window into the histogenesis of all Merkel cell carcinoma, with many authors regarding their cases as evidence that the cell of origin for all Merkel cell carcinoma is a primitive totipotent or multipotent cell capable of neuroendocrine, glandular, and/or squamous differentiation, whether localized to the epidermis, dermis, or follicular epithelium.8, 44, 50, 51, 52 The hypothesis of a common carcinogenic influence on two separate precursor cells39 has been suggested, as well as the theory of dedifferentiation of an already differentiated tumor type,11, 53 as exhibited by basal cell carcinoma54 and eccrine carcinoma,55 which may show neuroendocrine differentiation. Regardless of route of pathogenesis, the support for the inclusion of morphologic variants under the umbrella of Merkel cell carcinoma has come from electron microscopic and immunohistochemical evidence of neuroendocrine and epithelial differentiation within the small blue cell component. By using this evidence to define the commonality of these lesions, the obvious morphologic differences have been mostly disregarded.
Growing data support the likelihood that most squamous cell carcinomas/Merkel cell carcinomas are biologically distinct from pure Merkel cell carcinoma. Our findings of significant differences in protein expression and mutational patterns support this distinction. Recent findings from multiple authors have showed all combined Merkel cell carcinoma to be negative for Merkel cell polyomavirus by PCR and immunohistochemistry.1, 2, 13, 17, 19 Occasional reports document unusual features of these variants such as poor paranuclear globular staining with CK2047, 56, 57 and neurofilament negativity,56, 58 as seen in our cases, as well as ultrastructural and/or immunohistochemical absence of neurosecretory granules in the squamous components of the tumors.39, 41, 42, 44 Interestingly CK20 was diminished in two histopathologically undocumented cases reported by Bhatia, which had ‘lower viral abundance’.3 In an analysis of morphologic differences between Merkel cell polyomavirus-positive and -negative Merkel cell carcinoma, Kuwamoto’s group identified morphometrically reproducible variations in nuclear and cytoplasmic contour and content, where 4/6 of their Merkel cell polyomavirus-negative cohort were combined tumors.17 Unusual clinical backgrounds for combined—but not pure—Merkel cell carcinoma, include erythema ab igne,44, 59, 60 Marjolin’s ulcer8 and arsenical keratoses.11
Of late, several studies have examined a possible survival difference between Merkel cell polyomavirus+ and Merkel cell polyomavirus-Merkel cell carcinoma.4, 25, 27 Bhatia found distant metastases in 22% of viral abundant-RB1+ Merkel cell carcinoma compared with 43% of low virus-RB1-Merkel cell carcinoma, with a median survival difference of 86 vs 20 months (P<0.05).26 Waltari et al and Asioli et al found a negative disease-specific survival impact of tumor p53 expression.5, 30 Survival differences between combined and pure Merkel cell carcinoma have been inadequately addressed, but also invite consideration. Martin et al reported a high mortality rate in combined tumors (62%).1 Gomez reported an increased recurrence rate for squamous cell carcinoma/Merkel cell carcinoma compared with pure Merkel cell carcinoma (62 vs 26%), but no survival difference.39 Tang and Toker reported a difference in survival between ‘pure’ trabecular CA and combined squamous cell carcinoma/small cell tumors.61 In contrast, Walsh et al found no difference in outcome between epithelial and pure types,8 and Saeb-Lima62 showed better outcomes for Merkel cell carcinoma with eccrine and squamous differentiation. Our project was not intended as a case–control study, so we make no generalizations about survival. However, further study of outcomes data comparing these morphologic subsets, as better defined by immunohistochemical profiles, should follow, to clarify this issue.
We assessed expression of p63, which has been previously correlated with p53, is reported present in 60% of Merkel cell carcinoma, and is associated with a poorer overall and disease-free survival.29, 31, 32 In our cases all tumors (100%) were positive in the squamous cell carcinoma component, but only 60% in the NE component. We also assessed P53, and found that all of our cases were strongly and diffusely P53-positive in squamous cell carcinoma or both components. However, our length of follow-up is insufficient to compare survival with others (Table 2).
The possibility that the pathogenesis of Merkel cell carcinoma, particularly Merkel cell polyomavirus-negative Merkel cell carcinoma, is in part related to UV mutagenesis, has been suggested elsewhere.13, 16, 18, 63 There is a preferential geographical distribution of Merkel cell polyomavirus tumors to sun-exposed climates.16 Moreover, UV-associated mutations in the tumor suppressor p53 have been documented in some cases of Merkel cell carcinoma.63, 64 Increased P53 protein detection seems to be more commonly found in Merkel cell polyomavirus-negative tumors.4, 5 Similarly, pure squamous cell carcinoma, a tumor known to express P53, and thought to be pathogenetically related to p53 alterations, is the archetypal UV-related cutaneous carcinoma, and for the most part, does not express Merkel cell polyomavirus.65 Squamous cell carcinoma is well-known for its potential to dedifferentiate, suggesting the possibility that some combined tumors may be more closely related to squamous cell carcinoma, than to pure Merkel cell carcinoma.
Given our new knowledge of differential expression of antibodies to Merkel cell polyomavirus, and differential integration of viral DNA in morphologically distinct Merkel cell carcinoma,13, 17, 19, 66 we saw a reason to investigate the nature of these combined squamous and neuroendocrine tumors. All of our patients’ tumors presented on the sun-exposed area of the head and neck/or trunk of Caucasian persons, equating to ‘at-risk’ sites for UV-mediated damage, and contained varying degrees of neuroendocrine and squamous cell morphology. The identification of a shared P53+, RB1–, neurofilament– immunohistochemical profile, and a highly mutated, p53 and RB1 mutated molecular profile with a high rate of C>T mutations observed across the entire genomic target consistent with an UV light signature, different from pure Merkel cell carcinoma in all of these tumors supports that these tumors are biologically discrete entities. We therefore propose a classification system in which the term ‘Merkel cell carcinoma’ refers to histologically ‘pure’ Merkel cell polyomavirus+ tumors, whereas combined tumors are reported descriptively, such as primary cutaneous combined squamous cell (or basal cell or adnexal) and neuroendocrine carcinoma. Further studies are needed to investigate potential clinical relevance of these different categories. Regarding Merkel cell carcinoma', Toker once stated that ‘histogenetic considerations were, and always will be, secondary…and… essentially inconsequential’. Stern responded that, if there were a difference in origin, ‘is there a difference in prognosis…?’, and would a ‘histogenetic understanding … result in benefits to patients…?’.53 Accurate characterization of the phenotypic differences between the various subsets of primary cutaneous neuroendocrine carcinomas is the first step in determining if such distinctions will hold clinical importance.
References
Martin B, Poblet E, Rios JJ et al. Merkel cell carcinoma with divergent differentiation: histopathological and immunohistochemical study of 15 cases with PCR analysis for Merkel cell polyomavirus. Histopathology 2013;62:711–722.
Iwasaki T, Matsushita M, Kuwamoto S et al. Usefulness of significant morphologic characteristics in distinguishing between Merkel cell polyomavirus-positive and Merkel cell polyomavirus-negative Merkel cell carcinomas. Hum Pathol 2013;44:1912–1917.
Bhatia K, Goedert JJ, Modali R et al. Merkel cell carcinoma subgroups by Merkel cell polyomavirus DNA relative abundance and oncogene expression. Int J Cancer 2010;126:2240–2246.
Sihto H, Kukko H, Koljonen V et al. Merkel cell polyomavirus infection, large T antigen, retinoblastoma 1 protein and outcome in Merkel cell carcinoma. Clin Cancer Res 2011;17:4806–4813.
Waltari M, Sihto H, Kukko H et al. Association of Merkel cell polyomavirus infection with tumor p53, KIT, stem cell factor, PDGFR-alpha and survival in Merkel cell carcinoma. Int J Cancer 2011;129:619–628.
Skelton HG, Smith KJ, Hitchcock CL et al. Merkel cell carcinoma: analysis of clinical, histologic, and immunohistologic features of 132 cases with relation to survival. J Am Acad Dermatol 1997;37:734–739.
Llombart B, Monteagudo C, Lopez-Guerrero JA et al. Clinicopathological and immunohistochemical analysis of 20 cases of Merkel cell carcinoma in search of prognostic markers. Histopathology 2005;46:622–634.
Walsh NM . Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol 2001;32:680–689.
McCalmont TH . Paranuclear dots of neurofilament reliably identify Merkel cell carcinoma. J Cutan Pathol 2010;37:821–823.
Miettinen M . Synaptophysin and neurofilament proteins as markers for neuroendocrine tumors. Arch Pathol Lab Med 1987;111:813–818.
Visscher D, Cooper PH, ZaRB1o RJ et al. Cutaneous neuroendocrine (Merkel cell) carcinoma: an immunophenotypic, clinicopathologic, and flow cytometric study. Mod Pathol 1989;2:331–338.
Becker JC, Houben R, Ugurel S et al. MC polyomavirus is frequently present in Merkel cell carcinoma of European patients. J Invest Dermatol 2009;129:248–250.
Busam KJ, Jungbluth AA, Rekthman N et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol 2009;33:1378–1385.
Duncavage EJ, Zehnbauer BA, Pfeifer JD . Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol 2009;22:516–521.
Feng H, Shuda M, Chang Y et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008;319:1096–1100.
Garneski KM, Warcola AH, Feng Q et al. Merkel cell polyomavirus is more frequently present in North American than Australian Merkel cell carcinoma tumors. J Invest Dermatol 2009;129:246–248.
Kuwamoto S, Higaki H, Kanai K et al. Association of Merkel cell polyomavirus infection with morphologic differences in Merkel cell carcinoma. Hum Pathol 2011;42:632–640.
Ly TY, Walsh NM, Pasternak S . The spectrum of Merkel cell polyomavirus expression in Merkel cell carcinoma, in a variety of cutaneous neoplasms, and in neuroendocrine carcinomas from different anatomical sites. Hum Pathol 2012;43:557–566.
Paik JY, Hall G, Clarkson A et al. Immunohistochemistry for Merkel cell polyomavirus is highly specific but not sensitive for the diagnosis of Merkel cell carcinoma in the Australian population. Hum Pathol 2011;42:1385–1390.
Touze A, Gaitan J, Maruani A et al. Merkel cell polyomavirus strains in patients with merkel cell carcinoma. Emerg Infect Dis 2009;15:960–962.
Varga E, Kiss M, Szabo K et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol 2009;161:930–932.
Katano H, Ito H, Suzuki Y et al. Detection of Merkel cell polyomavirus in Merkel cell carcinoma and Kaposi's sarcoma. J Med Virol 2009;81:1951–1958.
Andres C, Belloni B, Puchta U et al. Prevalence of MCPyV in Merkel cell carcinoma and non-Merkel cell carcinoma tumors. J Cutan Pathol 2010;37:28–34.
Higaki-Mori H, Kuwamoto S, Iwasaki T et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol 2012;43:2282–2291.
Sihto H, Kukko H, Koljonen V et al. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst 2009;101:938–945.
Bhatia K, Goedert JJ, Modali R et al. Immunological detection of viral large T antigen identifies a subset of Merkel cell carcinoma tumors with higher viral abundance and better clinical outcome. Int J Cancer 2010;127:1493–1496.
Schrama D, Peitsch WK, Zapatka M et al. Merkel cell polyomavirus status is not associated with clinical course of Merkel cell carcinoma. J Invest Dermatol 2011;131:1631–1638.
Tan BH, Busam KJ, Pulitzer MP . Combined intraepidermal neuroendocrine (Merkel cell) and squamous cell carcinoma in situ with CM2B4 negativity and p53 overexpression(*). J Cutan Pathol 2012;39:626–630.
Asioli S, Righi A, Volante M et al. p63 expression as a new prognostic marker in Merkel cell carcinoma. Cancer 2007;110:640–647.
Asioli S, Righi A, de Biase D et al. Expression of p63 is the sole independent marker of aggressiveness in localised (stage I-II) Merkel cell carcinomas. Mod Pathol 2011;24:1451–1461.
Fleming KE, Ly TY, Pasternak S et al. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: report on a Canadian cohort. Hum Pathol 2014;45:952–960.
Hall BJ, Pincus LB, Yu SS et al. Immunohistochemical prognostication of Merkel cell carcinoma: p63 expression but not polyomavirus status correlates with outcome. J Cutan Pathol 2012;39:911–917.
Catalanotti F, Solit DB, Pulitzer MP et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res 2013;19:2257–2264.
Li H, DuRB1in R . Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–595.
DePristo MA, Banks E, Poplin R et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498.
Cibulskis K, Lawrence MS, Carter SL et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat biotechnol 2013;31:213–219.
Robinson JT, Thorvaldsdottir H, Winckler W et al. Integrative genomics viewer. Nat biotechnol 2011;29:24–26.
Tang CK, Toker C . Trabecular carcinoma of the skin: an ultrastructural study. Cancer 1978;42:2311–2321.
Gomez LG, DiMaio S, Silva EG et al. Association between neuroendocrine (Merkel cell) carcinoma and squamous carcinoma of the skin. Am J Surg Pathol 1983;7:171–177.
Sidhu GS, Feiner H, Flotte TJ et al. Merkel cell neoplasms. Histology, electron microscopy, biology, and histogenesis. Am J Dermatopathol 1980;2:101–119.
Tang CK, Toker C, Nedwich A et al. Unusual cutaneous carcinoma with features of small cell (oat cell-like) and squamous cell carcinomas. A variant of malignant Merkel cell neoplasm. Am J Dermatopathol 1982;4:537–548.
Al-Ahmadie HA, Mutasim DF, Mutema GK . A case of intraepidermal Merkel cell carcinoma within squamous cell carcinoma in-situ: Merkel cell carcinoma in-situ? Am J Dermatopathol 2004;26:230–233.
Ball NJ, Tanhuanco-Kho G . Merkel cell carcinoma frequently shows histologic features of basal cell carcinoma: a study of 30 cases. J Cutan Pathol 2007;34:612–619.
Iacocca MV, Abernethy JL, Stefanato CM et al. Mixed Merkel cell carcinoma and squamous cell carcinoma of the skin. J Am Acad Dermatol 1998;39:882–887.
Mott RT, Smoller BR, Morgan MB . Merkel cell carcinoma: a clinicopathologic study with prognostic implications. J Cutan Pathol 2004;31:217–223.
Rocamora A, Badia N, Vives R et al. Epidermotropic primary neuroendocrine (Merkel cell) carcinoma of the skin with Pautrier-like microabscesses. Report of three cases and review of the literature. J Am Acad Dermatol 1987;16:1163–1168.
Smith KJ, Skelton HG 3rd, Holland TT et al. Neuroendocrine (Merkel cell) carcinoma with an intraepidermal component. Am J Dermatopathol 1993;15:528–533.
Assaf C, Gellrich S, Whittaker S et al. CD56-positive haematological neoplasms of the skin: a multicentre study of the Cutaneous Lymphoma Project Group of the European Organisation for Research and Treatment of Cancer. J Clin Pathol 2007;60:981–989.
Adhikari LA, McCalmont TH, Folpe AL . Merkel cell carcinoma with heterologous rhabdomyoblastic differentiation: the role of immunohistochemistry for Merkel cell polyomavirus large T-antigen in confirmation. J Cutan Pathol 2012;39:47–51.
Gould E, Albores-Saavedra J, Dubner B et al. Eccrine and squamous differentiation in Merkel cell carcinoma. An immunohistochemical study. Am J Surg Pathol 1988;12:768–772.
Yamamoto O, Tanimoto A, Yasuda H et al. A combined occurrence of neuroendocrine carcinoma of the skin and a benign appendageal neoplasm. J Cutan Pathol 1993;20:173–176.
Kroll MH, Toker C . Trabecular carcinoma of the skin: further clinicopathologic and morphologic study. Arch Pathol Lab Med 1982;106:404–408.
Stern JB . "Murky cell" carcinoma (formerly trabecular carcinoma). A reply in which yet another name is revealed at the very end. Am J Dermatopathol 1982;4:517–519.
George E, Swanson PE, Wick MR . Neuroendocrine differentiation in basal cell carcinoma. An immunohistochemical study. Am J Dermatopathol 1989;11:131–135.
Banerjee SS, Eyden BP . Neuroendocrine differentiation in eccrine carcinoma. Histopathology 1996;29:389–390.
Brown HA, Sawyer DM, Woo T . Intraepidermal Merkel cell carcinoma with no dermal involvement. Am J Dermatopathol 2000;22:65–69.
Ferringer T, Rogers HC, Metcalf JS . Merkel cell carcinoma in situ. J Cutan Pathol 2005;32:162–165.
Hashimoto K, Lee MW, D'Annunzio DR et al. Pagetoid Merkel cell carcinoma: epidermal origin of the tumor. J Cutan Pathol 1998;25:572–579.
Jones CS, Tyring SK, Lee PC et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol 1988;124:110–113.
Hewitt JB, Sherif A, Kerr KM et al. Merkel cell and squamous cell carcinomas arising in erythema ab igne. Br J Dermatol 1993;128:591–592.
Toker C . Trabecular carcinoma of the skin. A question of title. Am J Dermatopathol 1982;4:497–500.
Saeb-Lima M, Montante-Montes de Oca D, Albores-Saavedra J . Merkel cell carcinoma with eccrine differentiation: a clinicopathologic study of 7 cases. Ann diagn pathol 2008;12:410–414.
Popp S, Waltering S, HeRB1st C et al. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int J Cancer 2002;99:352–360.
Van Gele M, Kaghad M, Leonard JH et al. Mutation analysis of P73 and TP53 in Merkel cell carcinoma. Br J Cancer 2000;82:823–826.
Reisinger DM, Shiffer JD, Cognetta AB Jr et al. Lack of evidence for basal or squamous cell carcinoma infection with Merkel cell polyomavirus in immunocompetent patients with Merkel cell carcinoma. J Am Acad Dermatol 2010;63:400–403.
Koba S, Inoue T, Okawa T et al. Merkel cell carcinoma with cytokeratin 20-negative and thyroid transcription factor-1-positive immunostaining admixed with squamous cell carcinoma. J Dermatol Sci 2011;64:77–79.
Acknowledgements
We are grateful for the support and assistance of the MSK Genomics Core Lab for assistance with Illumina sequencing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest. Research reported in this publication was supported by The Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Rights and permissions
About this article

Cite this article
Pulitzer, M., Brannon, A., Berger, M. et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol 28, 1023–1032 (2015). https://doi.org/10.1038/modpathol.2015.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.60
This article is cited by
-
RB1-deficient squamous cell carcinoma: the proposed source of combined Merkel cell carcinoma
Modern Pathology (2022)
-
Morphologic and immunophenotypical features distinguishing Merkel cell polyomavirus-positive and negative Merkel cell carcinoma
Modern Pathology (2019)
-
Are there multiple cells of origin of Merkel cell carcinoma?
Oncogene (2018)
-
Update on Merkel Cell Carcinoma
Head and Neck Pathology (2018)
-
Detection of the Merkel cell polyomavirus in the neuroendocrine component of combined Merkel cell carcinoma
Virchows Archiv (2018)
