
Abstract
The biological relevance of histological subtyping of ampullary carcinoma into intestinal vs pancreaticobiliary types remains to be determined. In an effort to molecularly profile these subtypes of ampullary carcinomas, we conducted a two-phase study. In the discovery phase, we identified 18 pancreatobiliary-type ampullary carcinomas and 14 intestinal-type ampullary carcinomas using stringent pathologic criteria and performed next-generation sequencing targeting 279 cancer-associated genes on these tumors. Although the results showed overlapping of genomic alterations between the two subtypes, trends including more frequent KRAS alterations in pancreatobiliary-type ampullary carcinoma (61 vs 29%) and more frequent mutations in APC in intestinal-type ampullary carcinoma (43 vs 17%) were observed. Of the entire cohort of 32 tumors, the most frequently mutated gene was TP53 (n=17); the most frequently amplified gene was ERBB2 (n=5); and the most frequently deleted gene was CDKN2A (n=6). In the second phase of the study, we aimed at validating our observation on ERBB2 and assessed ERBB2 amplification and protein overexpression in a series of 100 ampullary carcinomas. We found that (1) gene amplification and immunohistochemical overexpression of ERBB2 occurred in 13% of all ampullary carcinomas, therefore providing a potential target for anti-HER2 therapy in these tumors; (2) amplification and immunohistochemical expression correlated in all cases, thus indicating that immunohistochemistry could be used to screen tumors; and (3) none of the 14 ERBB2-amplified tumors harbored any downstream driver mutations in KRAS/NRAS, whereas 56% of the cases negative for ERBB2 amplification did, an observation clinically pertinent as downstream mutations may cause primary resistance to inhibition of EGFR family members.
Similar content being viewed by others


Main
Ampullary adenocarcinoma is a rare and heterogeneous malignancy occurring in 0.7 per 10 000 males and 0.4 per 10 000 females in the United States annually.1 Prognosis is generally dismal, with 5-year survival ranging from 4% in patients with distant metastases to 45% in stage 1 patients (SEER data).1 Because it forms at the junction of intestinal-type duodenal and pancreatobiliary-type ductal epithelium, ampullary carcinoma can have heterogeneous differentiation reflecting either or both of these types.2, 3 It has recently been shown that subtyping based on morphology, immunohistochemistry, and mRNA levels affects prognosis: patients with intestinal-type ampullary adenocarcinoma have a longer median overall survival of 70 months in comparison with the pancreatobiliary-type ampullary adenocarcinoma group, which has a median overall survival of 28 months.4, 5, 6
Recently, some oncologists have started treating ampullary carcinoma based on histologic subtype, using gemcitabine-based regimen for pancreatobiliary type and fluorouracil-based regimen for intestinal type. However, ∼12% of cases have mixed intestinal and pancreatobiliary differentiation2 and cannot be subtyped definitively into one category. Whether or not histologic subtype and cases with mixed differentiation have specific genetic signatures, and the influence of those signatures on prognosis and treatment response, remains to be investigated. In this study, we aimed to assess (1) whether histologic subtype correlated with differences in the somatic mutational and copy number profiles of 279 cancer-related genes; and (2) the targetable alterations that occur most frequently in ampullary carcinoma and their clinicopathologic and molecular correlates.
Materials and methods
Case Selection
After approval from our institutional review board, a discovery set and validation set were selected as follows. For the discovery set, unambiguous examples of 14 intestinal-type ampullary carcinomas and 18 pancreatobiliary-type ampullary carcinomas with matched normal tissues were selected for next-generation sequencing. Determination of intestinal vs pancreatobiliary subtype was performed on the basis of morphology and immunohistochemistry including expression of CDX2, CK7, CK20, MUC1, and MUC2.2 Tissue microarrays were constructed to validate the ERBB2 amplification finding discovered in the next-generation sequencing cases. For the validation set, all available institutional resection specimens from 1985 to 2013 were included. In total, 42 intestinal-type ampullary carcinomas, 44 pancreatobiliary-type ampullary carcinomas, 19 mixed intestinal and pancreatobiliary ampullary carcinomas, and one poorly differentiated ampullary carcinoma were studied. All cases tested by next-generation sequencing were also included in the tissue microarrays for correlation between methodologies, provided that adequate material was available.
Mutation Analysis
After macrodissection, genomic DNA was extracted from formalin-fixed paraffin-embedded tissue with the DNeasy Tissue KIT (Qiagen, Valencia, CA, USA). Next-generation sequencing of the discovery set was performed with the clinically validated next-generation sequencing assay, Integrated Mutation Profiling of Actionable Cancer Targets (IMPACT). This assay is a customized hybrid capture-based deep sequencing assay that evaluates 279 cancer-associated genes, listed in Supplementary Table 1. Detectable alterations include single-nucleotide variants, indels, and somatic copy number gains and losses. In brief, DNA was subjected to shearing, followed by library preparation. Matched normal tissue was processed in the same manner and samples were pooled together and sequenced on an Illumina HiSeq 2500. Burrows–Wheeler Aligner7 was used to align 100-bp paired end sequence reads to reference human genome. Single-nucleotide variants were detected using MuTect,8 whereas small indels were identified using SomaticIndelDetector. Germline variants were filtered out based on the matched germline DNA.
Manual review was performed using IntegratedGenomicsViewer for all candidate mutations.9 Technical details of this assay are further described elsewhere.10
KRAS, BRAF, NRAS, and PIK3CA mutation testing was performed on all ERBB2-amplified cases discovered from the tissue microarrays that did not undergo IMPACT testing. The methodology used for this further testing was the MassARRAY system (Sequenom) with primers as previously described11, 12 at the following hotspots in duplicate for KRAS: c.34, 35, 37, 38, 181, 182, 183, 351, and 437; NRAS c. 34, 35, 37, 38, 181, 182, and 183; BRAF c. 1781, 1798 and 1799; and PIK3CA c. 1624, 1633, and 3140.
ERBB2 Analysis with Whole Sections and Tissue Microarrays
All cases that underwent IMPACT analysis from the discovery set were immunohistochemically stained for ERBB2 expression on whole sections. In addition, three 0.6-mm-diameter cores from separate areas of all available ampullary carcinoma cases were used to construct tissue microarrays. Duplicate tissue microarrays were constructed to account for core dropout. Immunohistochemistry was performed using PATHWAY anti- HER-2/neu (4B5) Rabbit Monoclonal Primary Antibody (Ventana Medical Systems; Tucson, AZ, USA). Immunohistochemical analysis paralleled that used for gastric carcinoma.13 Basolateral and/or complete membranous staining were scored, whereas cytoplasmic and nuclear staining, as well as staining of nontumor elements, were not included. A cluster of at least five positive tumor cells was required, and immunohistochemical staining intensity was interpreted using magnification necessary to assess staining: 3+ intensity was visible at × 40 original magnification, 2+ intensity was visible at × 100– × 200 original magnification, 1+ intensity was visible at × 400 original magnification, and 0 intensity was used in the absence of staining.
Chromogenic in situ hybridization was performed using INFORM HER2 Dual ISH DNA Probe Cocktail Assay (Ventana Medical Systems). Twenty tumor cells per core were counted. An ERBB2: CEP17 ratio of >2 in one core was considered amplified, as were clusters of ERBB2 signals that could not be counted individually owing to overlap of many signals.
Results
IMPACT Findings
Among 32 cases tested by IMPACT, mean target coverage of normal and tumor DNA was 327 × and 412 × , respectively. Aside from one hypermutated case with 225 mutations, which was negative for ERBB2 amplification, the number of mutations per tumor ranged from 1 to 16, with an average of five mutations per ampullary carcinoma case, excluding the single hypermutator case. Copy number alterations ranged from 0 to 6, with an average of one copy number alteration per tumor.
Histologic Subtype Trends for Common Mutations Seen in Colorectal and Pancreatic Carcinoma
Trends between molecular alterations and phenotype included more frequent KRAS alterations in pancreatobiliary-type ampullary carcinoma than intestinal-type ampullary carcinoma (61 vs 29%) and more frequent mutations in APC in intestinal-type ampullary carcinoma than pancreatobiliary-type ampullary carcinoma (43 vs 17%). CDKN2A was more frequently deleted in intestinal-type ampullary carcinoma than in pancreatobiliary-type ampullary carcinoma (30 vs 6%). However, these trends did not reach statistical significance (P>0.05).
Approximately 13% of Ampullary Carcinomas Exhibit ERBB2 Amplification, without Predilection for Subtype
Combining data from IMPACT and chromogenic in situ hybridization, 14 of 106 cases (13%) showed ERBB2 amplification. The clinicopathologic features of all ampullary carcinoma tested by IMPACT or chromogenic in situ hybridization/immunohistochemistry for ERBB2 are summarized in Table 1. No significant differences in age, sex, subtype, node status, peri-neural, or lymphovascular invasion were identified between ERBB2-amplified and nonamplified ampullary carcinoma. Interestingly, several cases had missense mutations in ERBB2. All ERBB2 missense mutations occurred in KRAS/BRAF wild type. Two cases with ERBB2 mutations occurred in ERBB2-amplified ampullary carcinoma (Table 2), whereas other ERBB2 mutations in three nonamplified ampullary carcinoma included ERBB2 p. R678Q, R103L, and R784C. ERBB2 missense mutation did not correlate with histologic subtype.
ERBB2 Amplification Frequently Co-occurs with TP53 Mutation but Not KRAS, NRAS, or BRAF Mutation
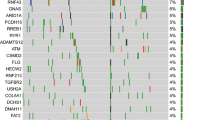
The most frequent gains (including five cases of ERBB2 amplification), losses, and point mutations and indels in the discovery set are summarized in Figure 1. The molecular profiles of the ampullary carcinoma cases with ERBB2 amplification are summarized in Table 2. None of the 14 ERBB2-amplified cases had KRAS or NRAS c. 12, 13, 61, or 117, whereas 15 of 27 (56%) of cases without ERBB2 amplification were positive for a KRAS or NRAS c. 12, 13, 61, and 117 mutations. However, one ERBB2-amplified case (detected via chromogenic in situ hybridization) was positive for a PIK3CA p. H1047R (c. 3140A>G) mutation on Sequenom analysis. Interestingly, all five cases with ERBB2 amplification on IMPACT had concomitant TP53 mutations, whereas only 44% of ampullary carcinoma cases without ERBB2 amplification had TP53 mutations.

Most frequent genetic alterations detected via IMPACT. (a) ERBB2 and neighboring gene CDK12 were the most frequently amplified genes, whereas (b) CDKN2A was the most frequently deleted. (c) TP53 was the most frequent of the missense/indel mutations followed by KRAS and APC, which each respectively occurred more frequently in pancreatobiliary and intestinal-type ampullary carcinoma.
ERBB2 Immunohistochemistry Results Demonstrate High Sensitivity for Amplification by Chromogenic In Situ Hybridization and IMPACT
All five cases that were amplified on IMPACT displayed strong, diffuse immunohistochemical positivity on whole sections. Among the 29 cases that were negative for ERBB2 amplification on IMPACT, 6 cases displayed heterogeneity on whole sections including 4 cases ranging from 0 to 2+ in intensity, 1 case that ranged from 1+ to 2+ in intensity, and 1 case that ranged from 0 to 1+ in intensity.
Among the 100 cases available for chromogenic in situ hybridization analysis, 13 were positive for ERBB2 amplification, and the correlation between immunohistochemistry intensity and chromogenic in situ hybridization result is shown in Table 1. Immunohistochemical staining detected all 13 cases with ERBB2 amplification, yielding a sensitivity of 100%. Furthermore, lack of 3+ immunohistochemical intensity predicted lack of ERBB2 amplification in all 87 cases without ERBB2 amplification, yielding a specificity of 100% (Figure 2). There were no discrepancies between IMPACT and chromogenic in situ hybridization on cases tested by both methods, including 4 ERBB2-amplified cases and 22 ERBB2 nonamplified cases. Immunohistochemical heterogeneity between cores occurred in two amplified cases, yet on each of these two cases chromogenic in situ hybridization showed consistent homogeneous results between cores (Figure 2).

ERBB2 immunohistochemistry and chromogenic in situ hybridization correlation. (a) A pancreatobiliary-type ampullary carcinoma showing (b) strong membranous staining (3+) visible on low power and (c) clustering of numerous ERBB2 signals per cell (black dots) on chromogenic in situ hybridization, consistent with amplification. (d–h) In this case, (d) a more poorly differentiated area had (e) no immunohistochemical reactivity, yet (f) it was amplified on chromogenic in situ hybridization, whereas (g) a more well differentiated area with intestinal differentiation had (h) 2+ immunohistochemical reactivity and (i) similar amplification on CISH (a, d, g, h and e), × 100 original magnification; b, e, h: 4B5 immunohistochemistry, × 100 original magnification; C, F, I: ERBB2 CISH, × 1000 original magnification; both: Ventana Medical Systems; D and G: H&E, × 400 original magnification;e and h: 4B5 immunohistochemistry, × 400 original magnification).
Discussion
In this study, we have found that the incidences of mutations frequently implicated in colorectal and pancreatic carcinogenesis, in particular mutations in APC and KRAS, correlate with the specific histologic subtype of ampullary carcinoma. Although neither APC nor KRAS mutations were 100% specific for pancreatobiliary-type or intestinal-type ampullary carcinoma, respectively, it is noted that APC mutations do occur, though rarely, in pancreatic carcinoma. The differences identified in the genetic signatures of subtypes of ampullary carcinoma may help assess prognosis, as well as treatment regimen, and may help assist with classification when the histologic subtype is mixed. In addition, but not surprisingly, a number of other alterations in tumor suppressors and oncogenes occurred in both groups, including ERBB2 amplification. This is the first systematic investigation of ERBB2 amplification and overexpression in ampullary carcinoma and their molecular correlations. In addition to identifying the most frequently mutated oncogenes and tumor suppressors, we have shown that ERBB2 amplification is a relatively frequent event in ampullary carcinoma with specific molecular correlates and can be reliably identified using routine immunohistochemistry.
Our results correlate well with those of previous studies that have demonstrated ERBB2 amplification/overexpression in 0–23% of cases.14, 15, 16 In addition to previous studies, we provide data correlating ERBB2 in situ hybridization and immunohistochemical results in ampullary carcinoma. We have found that the immunohistochemical scoring criteria used for gastric carcinoma work well for ampullary carcinoma for predicting gene amplification. Further, the molecular profiles of ERBB2-amplified ampullary carcinoma included frequent TP53 co-mutation and a paucity of KRAS or NRAS mutations. Thus, ERBB2-amplified ampullary carcinomas have a molecular profile that is more similar to gastric than colorectal or pancreatobiliary carcinoma: provisional TCGA data show that pancreatobiliary carcinomas have a paucity of ERBB2 amplification, and colorectal carcinomas do not share the positive association between TP53 mutation and ERBB2 amplification or the negative correlation between KRAS/BRAF mutation and ERBB2 amplification.17 The fact that ERBB2-amplified ampullary carcinoma did not harbor downstream mutations in KRAS or BRAF may be important therapeutically, as the latter may cause primary resistance to inhibition of EGFR family members.18 With a similar molecular profile to ERBB2-amplified gastric carcinoma, it follows that ERBB2-amplified ampullary carcinoma might also derive benefit from similar targeted therapy. The significance of the ERBB2 mutations detected in this study is yet to be determined. None of these mutations occurred at hot spots; however, two mutations occurred in cases positive for ERBB2 amplification: the ERBB2 p. L313I extracellular domain mutation occurred in 95% of reads, suggesting that the mutation was amplified, although the ERBB2 p. D639V mutation occurred in only 5% of reads, suggesting that it occurred in a nonamplified subclone. The other ERBB2 mutations in three nonamplified cases (p. R678Q, R103L, and R784C) have not been described in the Cosmic database or other reports.
Intratumoral heterogeneity of ERBB2 amplification is relatively common in gastric carcinoma, yet not as common in breast carcinoma. We identified intra-tumoral heterogeneity in 2/14 (13%) of cases based on immunohistochemistry, yet not by chromogenic in situ hybridization. It is important to note that all three cores per tumor were derived from the same paraffin block, and whether the ERBB2 status of synchronous or metachronous metastases will correlate well with the ERBB2 status of the primary ampullary carcinoma remains to be seen. One possible cause of the discordant immunohistochemistry results in these two cases is less than optimal tissue fixation. Ampullary carcinoma would be especially prone to fixation issues, as these resections typically include pancreatic tissue with high levels of digestive enzymes. Another possible reason would be epigenetic changes altering protein expression, as both cases were phenotypically heterogeneous.
Limitations to this study include the relatively small numbers of histologically typical intestinal-type and pancreatobiliary-type ampullary carcinoma, as well as relatively small numbers of ERBB2-amplified ampullary carcinoma, which precluded meaningful survival and treatment response comparisons. Our methodology was limited to DNA-level alterations without specific assessment of possible epigenetic mechanisms. It has recently been shown that differences in phenotypic differentiation of ampullary carcinoma are reflected in RNA profiling,4 perhaps suggesting that epigenetic factors may additionally contribute to phenotypic differentiation. Regarding assessment of ERBB2 amplification, specific fixation requirements were not required for inclusion in this study; whether stricter fixation requirements would alter immunohistochemical patterns in ampullary carcinoma remains to be seen.
In summary, certain molecular trends may be associated with intestinal or pancreatobiliary histologic subtype in ampullary carcinoma, and these molecular trends may be important for therapeutic decision-making and prognosis. ERBB2 amplification occurs in ∼13% of ampullary carcinoma, is virtually mutually exclusive with downstream mutations in KRAS/NRAS/BRAF, and is present regardless of subtype or nodal status. ERBB2 amplification can be reliably screened for using immunohistochemistry with the scoring criteria developed for gastric carcinoma, and patients with ERBB2-amplified ampullary carcinoma may be candidates for targeted therapy.
References
Horner MJ, Ries LAG, Krapcho M et al. (eds). SEER Cancer Statistics Review, 1975–2006, National Cancer Institute, Bethesda, MD, USA http://seer.cancer.gov/csr/1975_2006/ based on November 2008 SEER data submission, posted to the SEER web site, 2009.
Ang DC, Shia J, Tang LH et al. The utility of immunohistochemistry in subtyping adenocarcinoma of the ampulla of vater. Am J Surg Pathol 2014;38:1371–1379.
Adsay V, Ohike N, Tajiri T et al. Ampullary region carcinomas: definition and site specific classification with delineation of four clinicopathologically and prognostically distinct subsets in an analysis of 249 cases. Am J Surg Pathol 2012;36:1592–1608.
Overman MJ, Zhang J, Kopetz S et al. Gene expression profiling of ampullary carcinomas classifies ampullary carcinomas into biliary-like and intestinal-like subtypes that are prognostic of outcome. PLoS One 2013;8:e65144.
Bronsert P, Kohler I, Werner M et al. Intestinal-type of differentiation predicts favourable overall survival: confirmatory clinicopathological analysis of 198 periampullary adenocarcinomas of pancreatic, biliary, ampullary and duodenal origin. BMC Cancer 2013;13:428.
Zhou H, Schaefer N, Wolff M et al. Carcinoma of the ampulla of Vater: comparative histologic/immunohistochemical classification and follow-up. Am J Surg Pathol 2004;28:875–882.
Li H, Durbin R . Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26:589–595.
Cibulskis K, Lawrence MS, Carter SL et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–219.
Robinson JT, Thorvaldsdóttir H, Winckler W et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–26.
Jelinic P, Mueller JJ, Olvera N et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet 2014;46:424–426.
Arcila M, Lau C, Nafa K et al. Detection of KRAS and BRAF mutations in colorectal carcinoma roles for high-sensitivity locked nucleic acid-PCR sequencing and broad-spectrum mass spectrometry genotyping. J Mol Diagn 2011;13:64–73.
Chaft JE, Arcila ME, Paik PK et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther 2012;1:485–491.
Hechtman JF, Polydorides AD . HER2/neu gene amplification and protein overexpression in gastric and gastroesophageal junction adenocarcinoma: a review of histopathology, diagnostic testing, and clinical implications. Arch Pathol Lab Med 2012;136:691–697.
Aloysius MM, Lobo DN, Rowlands BJ et al. HER-2/Neu overexpression is a rare event in peri-ampullary cancer: assessment using the HercepTest. Histopathology 2009;55:236–237.
Baumhoer D, Zlobec I, Tornillo L et al. Immunophenotyping and oncogene amplifications in tumors of the papilla of Vater. Virchows Arch 2008;453:579–588.
Ajiki T, Kamigaki T, Hasegawa Y et al. Proliferating cell nuclear antigen, p53, and c-erbB-2 expression in relation to clinicopathological variables and prognosis in cancer of the ampulla of Vater. Hepatogastroenterology 2001;48:1266–1270.
Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404.
Ramalingam SS, Blackhall F, Krzakowski M et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2012;30:3337–3344.
Acknowledgements
The MSKCC Sequenom facility was supported by the Anbinder Fund.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website .
Supplementary information
Rights and permissions
About this article

Cite this article
Hechtman, J., Liu, W., Sadowska, J. et al. Sequencing of 279 cancer genes in ampullary carcinoma reveals trends relating to histologic subtypes and frequent amplification and overexpression of ERBB2 (HER2). Mod Pathol 28, 1123–1129 (2015). https://doi.org/10.1038/modpathol.2015.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.57
This article is cited by
-
Optimizing Patient Pathways in Advanced Biliary Tract Cancers: Recent Advances and a French Perspective
Targeted Oncology (2023)
-
A novel hotspot and rare somatic mutation p.A138V, at TP53 is associated with poor survival of pancreatic ductal and periampullary adenocarcinoma patients
Molecular Medicine (2020)
-
Primary small bowel adenomas and adenocarcinomas—recent advances
Virchows Archiv (2018)
-
HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: a potential therapeutic target?
Cancer and Metastasis Reviews (2017)
-
Ampullary carcinoma is often of mixed or hybrid histologic type: an analysis of reproducibility and clinical relevance of classification as pancreatobiliary versus intestinal in 232 cases
Modern Pathology (2016)
