
Abstract
Desmoplastic malignant melanoma is a distinct melanoma entity histologically subtyped into mixed and pure forms due to significantly reduced lymph node metastases in the pure form. Recent reports investigating common actionable driver mutations have demonstrated a lack of BRAF, NRAS, and KIT mutation in pure desmoplastic melanoma. In search for alternative driver mutations next generation amplicon sequencing for hotspot mutations in 50 genes cardinal to tumorigenesis was performed and in addition the RET G691S polymorphism was investigated. Data from 21 desmoplastic melanomas (12 pure and 9 mixed) were retrieved. Pure desmoplastic melanomas were either devoid of mutations (50%) or displayed mutations in tumor suppressor genes (TP53, CDKN2A, and SMAD4) singularly or in combination with the exception of a PIK3CA double-mutation lacking established biological relevance. Mixed desmoplastic melanomas on the contrary were frequently mutated (89%), and 67% exhibited activating mutations similar to common-type cutaneous malignant melanomas (BRAF, NRAS, FGFR2, and ERBB2). Separate analysis of morphologically heterogeneous tumor areas in four mixed desmoplastic malignant melanomas displayed no difference in mutation status and RET G691 status. GNAQ and GNA11, two oncogenes in BRAF and NRAS wild-type uveal melanomas, were not mutated in our cohort. The RET G691S polymorphism was found in 25% of pure and 38% of mixed desmoplastic melanomas. Apart from RET G691S our findings demonstrate absence of activating driver mutations in pure desmoplastic melanoma beyond previously investigated oncogenes (BRAF, NRAS, and KIT). The findings underline the therapeutic dichotomy of mixed versus pure desmoplastic melanoma with regard to activating mutations primarily of the mitogen-activated protein kinase pathway.
Similar content being viewed by others


Main
Desmoplastic malignant melanoma is a rare subtype of spindle cell malignant melanoma composed of spindled tumor cells interspersed in a characteristic, dense, paucicellular fibrous stroma, or fibromyxoid stroma.1 A variable admixture of an epithelioid or nondesmoplastic-spindled tumor cell component resembling conventional malignant melanoma is frequently seen. Presence of a conventional (nondesmoplastic) malignant melanoma component of >10% has been used as a cut-off to define mixed desmoplastic malignant melanoma as opposed to pure desmoplastic malignant melanoma. Desmoplastic malignant melanomas account for <4% of cutaneous melanoma cases and patients tend to present with advanced stage of disease due to lack of lesional pigmentation and occasional initial misdiagnosis.2 Clinically, desmoplastic malignant melanoma is frequently associated with lentigo maligna. When compared to conventional malignant melanoma of identical thickness, desmoplastic malignant melanoma has been associated with a reduced rate of lymph node metastases but higher local recurrence rates, most likely reflecting incomplete excisions of these tumors.3, 4 In addition, desmoplastic malignant melanoma exhibits a propensity for hematogenous spread primarily to the lung.5 Histological subclassification into pure and mixed desmoplastic malignant melanoma has been advocated on grounds of a purportedly negligible risk for lymph node metastases in pure desmoplastic malignant melanoma and restriction to sentinel node biopsy to patients with clinically conspicuous lymph nodes.6, 7 Targeted therapies (vemurafenib and imatinib) available for patients harbouring BRAF,8 NRAS,9 and KIT10 mutations have added therapeutic relevance to mutational subtyping of malignant melanoma. Increasing evidence demonstrated that mutational characteristics segregate with currently accepted subtyping based on histology and tumor location:11, 12, 13, 14, 15, 16 uveal melanomas present the prototypical case of a melanoma entity with a distinct mutation profile as they typically lack BRAF mutations,17 but instead frequently display GNAQ or GNA11 mutations.18, 19 Spitzoid melanomas demonstrate a reduced BRAF mutation frequency compared to general melanoma,11 and mucosal melanomas are frequently KIT mutated but BRAF wild type.20 Regarding desmoplastic malignant melanoma a complete lack of BRAF mutations was reported in one publication21 and a markedly decreased frequency of BRAF mutations in another.22 Subclassification of desmoplastic malignant melanoma into pure and mixed type is not only clinically relevant, but also apparently reflects different mutation patterns as well. Miller et al23 have recently shown that pure desmoplastic malignant melanoma is devoid of BRAF mutations as opposed to mixed desmoplastic malignant melanoma, which did exhibit BRAF mutations albeit at a low level (6% of cases). In addition, absence of BRAF mutations in an investigation including 10 desmoplastic melanomas (not further subclassified) has been reported by another group.24 To identify other, yet unrecognized oncogenic mutations in desmoplastic malignant melanoma we investigated 23 desmoplastic malignant melanomas by next generation amplicon sequencing using a mutation panel (IonAmpliSeq Cancer Hotspot Panel v2, Life Technologies), which covers 2855 COSMIC-annotated hotspot mutations of 50 cardinal tumor-related genes (for genes and respective amplicons included, as well as their genomic coordinates see Supplementary Table 1). This panel includes among others BRAF, CDKN2A, BRAF, TP53, PTEN, KIT as well as GNAQ, and GNA11, the latter reported in BRAF wild type uveal melanomas, but to our knowledge not yet investigated in desmoplastic malignant melanoma.
The mutation panel also includes several mutation hotspots of the receptor tyrosine kinase RET, a protooncogene with an amino-acid changing single-nucleotide polymorphism in the juxtamembrane region of RET, namely G691S (RETp). This polymorphism enhances the response of RET to glial cell line-derived neurotrophic factor, which activates both the RET-RAF-RAS-MEK-ERK pathway and the RET-PIK3-AKT pathway.25, 26 This activation has been linked to the observed neurotropism in desmoplastic melanoma and pancreatic ductal adenocarcinoma among others. Narita et al25 have described a significantly higher frequency of RETp in desmoplastic melanomas (61%) compared to nondesmoplastic melanomas (31%) analyzing RETp from tumor DNA, but Miller et al23 have only detected RETp in 33% of pure and in 24% of mixed desmoplastic malignant melanoma. In the light of occasional, somatic, de novo RETp occurrences reported in pancreatic adenocarcinoma,27 Bar et al28 have investigated germline DNA for RETp in melanoma patients and found germline RETp in 30% of patients with desmoplastic melanomas, compared to 21% in nondesmoplastic malignant melanoma patients, but this difference was not significant at the investigated sample size. As RET codon 691 is not covered by the abovementioned hotspot panel, tumor DNA was subjected to Sanger sequencing for additional evaluation of this RET codon in order to encompass germline and possible somatic RET G691S occurrences.
Materials and Methods
Twenty-three patients with histologically confirmed desmoplastic malignant melanomas were included in the study. Formalin-fixed paraffin-embedded tumor tissue of the primary tumor was retrieved from the archives of the Biobank of the Medical University of Graz, the Dermatopathologie Bodensee, Friedrichshafen, Germany, the Department of Dermatology and Venereology of the Medical University of Graz and from Austrian district hospital pathologies in three consultation cases. H&E and immunohistochemical slides were reevaluated to confirm the diagnoses by two independent pathologists (BL-A and SWJ) prior to inclusion in the study. Immunohistochemically, all tumors were positive for S100. When stratified for the presence of a desmoplastic tumor component of at least 90%, 13 desmoplastic malignant melanoma were classified as pure and 10 tumors were classified as mixed desmoplastic malignant melanomas. See Figure 1 for exemplary tumor morphology of cases analyzed.

(a) Pure desmoplastic malignant melanoma with spindled tumor cells embedded in fibrotic stroma; inset (a) tumor cells with S100 reactivity (high power). (b) Mixed desmoplastic malignant melanoma with areas of spindle cells and desmoplastic stroma as well as nondesmoplastic areas with epithelioid tumor cells (inset (b), high power).
Serial sections with first and last levels H&E stained for topographic reference were obtained. Unstained, intermediate sections were mounted on glass slides for selective manual microdissection with a scalpel. DNA was extracted on a Maxwell, MDx Research System (Promega, Fitchburg, Wisconsin, USA). In cases with limited tumor tissue content laser microdissection on a Veritas microdissection instrument (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to standard protocols was performed and DNA was purified using the QIAmp DNA mini kit (Qiagen, Hilden, Germany). Primers for selected target regions of 50 tumor-associated genes (Table 1), corresponding to 2855 COSMIC-annotated hotspot mutations are included in the Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific). Library preparations were performed using the Ion AmpliSeq library kit 2.0 (Thermo Fisher Scientific). Sequencing was performed on an Ion PGM Sequencer (Thermo Fisher Scientific). Emulsion PCR and sequencing were performed with the appropriate kits (Ion One Touch Template Kit v2 and Ion PGM 200 sequencing kit, Thermo Fisher Scientific) multiplexing four or eight samples on 316 or 318 chips, respectively. Sequencing length was set to 500 flows and yielded reads ranging from 111 to 187 base pairs consistent with the expected size range; 350.000 to 500.000 reads were obtained for each sample with >80% of bases at AQ20.
Data Analysis
Initial data analysis was performed using the Ion Torrent Suite Software Plug-ins (Thermo Fisher Scientific, open source, GPL, https://github.com/iontorrent/). Briefly, this included base calling, alignment to the reference genome (HG19) using the TMAP mapper and variant calling by a modified diBayes approach taking into account the flow space information. All called variants were annotated using open source software (ANNOVAR, http://www.openbioinformatics.org/annovar/;29 SnpEff, http://snpeff.sourceforge.net/30) and custom Perl scripts. All coding, nonsynonymous mutations were further evaluated and visually inspected in IGV (http://www.broadinstitute.org/igv/) and variant calls resulting from technical read errors or sequence effects were excluded from the analysis. Mutations were subsequently assessed for biological relevance by computerized prediction inferred by a particular amino acid change. ‘Sorting intolerant from tolerant’ algorithm (SIFT) was used to generate SIFT-scores.31 Amino acid substitutions with scores below 0.05 were interpreted as functionally relevant (prediction ‘damaging’), scores between 0.05 and 0.1 were regarded as possibly relevant (‘possibly damaging’) and scores >0.1 were regarded as irrelevant (‘functionally neutral’) by computerized prediction. Final assessment of biological relevance was interpreted in conjunction with data from the COSMIC database32 (accessed November 2014) and literature search as explained in the text.
Sequence analysis for evaluation of codon 691 of RET exon 11 by Sanger sequencing was performed according to standard procedures from forward and reverse primer. PCR reactions were carried out in a final volume of 20 μl with the HotStarTaq Plus Master Mix kit (Qiagen, Hilden, Germany), 0.4 μmol/l primers (F-5′-GTCTGCCTTCTGCATCCACT-3′, R-5′-CCTCACCAGGATCTTGAAGG-3′; VBC-Biotech Service GmbH, Vienna, Austria), PCR-grade water (Roche Diagnostics, Mannheim, Germany) and 25 ng tumor DNA. For purification and sequencing PCR products were sent to the VBC-Biotech Service GmbH. Sequence analysis was performed with the CodonCode Aligner sequence analysis software (CodonCode Corporation, Centerville, MA, USA).
SPSS Statistics 22, IBM, was used to calculate Fisher’s exact tests for statistical analyses of RETp and mutation frequencies between pure and mixed desmoplastic malignant melanomas.
Results
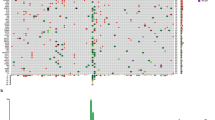
Twenty-three patients were included in the study. After generation of amplicon libraries, 21 of the 23 samples yielded sequencing raw data suitable for further analysis (12 pure and 9 mixed desmoplastic malignant melanoma from 14 male and 7 female patients). Two of the three laser microdissected samples yielded sequencing data unsuitable for further evaluation due to incomplete amplicon coverage and were excluded from further analysis. Tumors were located at the upper torso/arm in nine cases, in the face/at the head in eight cases and at the neck/shoulder in four cases. Median age at tumor presentation was 72 years (mean 70.9 years). Table 2 details histological type of desmoplastic malignant melanoma (pure/mixed), sex, patient age, tumor location, mutations detected, RETp status, presence of perineural invasion, histological evidence of UV damage and concomitant in situ lesions. All tumors were at least TNM stage pT4 with the exception of one case (case 12 in Table 2) that was pT3a. Nonsynonymous (amino acid changing) mutations were found in 14 out of 21 desmoplastic malignant melanomas (67%) with a total of 25 mutations detected (see Table 3 for details). All mutations were point mutations, comprising three stopgain mutations in CDKN2A and TP53. When subclassified into pure and mixed desmoplastic malignant melanoma 6 out of 12 (50%) pure desmoplastic malignant melanomas were devoid of mutations in our analysis. Pure desmoplastic malignant melanomas with mutations exhibited mutated tumor suppressor genes in five out of six cases mainly affecting TP53 and CDKN2A as well as SMAD4 in one case. Only one pure desmoplastic malignant melanoma displayed a mutated protooncogene (PIK3CA). In contrast, all but one mixed desmoplastic malignant melanoma were mutated with oncogenic mutations in two-thirds of cases. The observed difference of oncogenic mutations between pure and mixed desmoplastic malignant melanomas is statistically significant at P=0.016 by Fisher’s exact test and this difference is even more pronounced (P=0.0015) under the assumption of the one PIK3CA double-mutation detected in pure desmoplastic melanoma being in fact a functionally irrelevant passenger mutation as explicated in the discussion. Activating mutations were found in BRAF (V600E and V600R), in NRAS, FGFR2 (twice), and in ERBB2. Tumor suppressor gene mutations pertained to TP53 in 4, CDKN2A in 1, SMARCB1 in 1, and to PTPN11 in 1 out of 9 mixed desmoplastic malignant melanomas. None of the tumors analyzed exhibited mutations in KIT, GNAQ, or GNA11. RET codon 691 status was retrieved in all but one case, which even on increased DNA input failed to amplify. Overall, RETp was seen in 30% of melanomas. RETp frequency was 25% for pure desmoplastic malignant melanomas and 38% for mixed desmoplastic malignant melanomas. Perineural invasion could be assessed in 19 out of 21 cases, while in two cases not enough peritumoral tissue was resected for evaluation. Seven tumors demonstrated perineural invasion of which three (43%) were RETp. Twelve cases lacked perineural invasion of which again three (25%) were RETp. No statistically significant difference was found between RETp frequencies of pure versus mixed desmoplastic malignant melanomas (P=NS) at the given sample size.
Discussion
Malignant melanoma is currently subtyped based on location and histomorphology. Analysis for mutations currently amenable to therapy (ie, BRAF, NRAS, and KIT) have revealed mutational patterns segregating with the traditional subgroups. Regarding desmoplastic malignant melanoma a complete absence of BRAF mutations21 as well as a reduced mutation rate22 has been reported. Recently, Miller et al23 have observed a lack of BRAF mutations in pure desmoplastic malignant melanoma compared to a low frequency in the mixed subtype and a trend toward an increased RETp frequency in pure desmoplastic malignant melanoma. Kim et al24 extended mutation analysis in desmoplastic malignant melanoma to BRAF and KIT reporting wild-type BRAF and KIT in all tumors investigated.
Evidence that activating BRAF and NRAS mutations do not represent the main driver mutations in desmoplastic malignant melanoma prompted us to search for other, yet unaccounted mutations that might be responsible for tumor development and potentially be amenable to therapeutic intervention. Data from 21 desmoplastic malignant melanomas (12 pure and 9 mixed) were retrieved by next generation amplicon sequencing for hotspot mutations in 50 cancer-related genes employing the IonAmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific).
Two-thirds (67%) of pure desmoplastic malignant melanomas were devoid of mutations. Within the group of pure desmoplastic malignant melanomas only a single case demonstrated a mutated protooncogene (PIK3CA). The remaining pure DMMs displayed mutations in tumor suppressor genes only. The activating mutation present in that case was a PIK3CA double mutation, of which we regard one as an unequivocal passenger mutation (p.F83L, no COSMIC entries, SIFT score 0.6), while the other one (p.N345S, SIFT score 0.09—‘possibly damaging’) we view to be likely nonpathogenic as well, with only two COSMIC entries unconfirmed for somatic occurrence together with the fact that PIK3CA is a widely investigated gene. The remaining five pure desmoplastic malignant melanomas mutated displayed mutations in two recurrent tumor suppressor genes namely TP53 and CDKN2A. The SMAD4 mutation in case 4 appears to be a passenger mutation without biological relevance (no COSMIC entries, SIFT score 0.16). As RETp could only account for proliferative activation in 38% of pure desmoplastic malignant melanomas in our series, the absence of canonical activating mutations, even after analysis with an extended multigene tumor panel, points to a still elusive proliferative stimulus in pure desmoplastic malignant melanoma. Irrespective of the activating pathogenic alterations driving these tumors, mutations in well-known tumor suppressor genes could be identified as relevant to neoplastic transformation in ~40% of pure desmoplastic malignant melanomas.
Results in mixed desmoplastic malignant melanoma were markedly different. All but one mixed desmoplastic malignant melanoma (89%) were mutated, frequently displaying oncogenic mutations of a spectrum similar to that seen in melanoma in general (Tables 2 and 3). The activating mutation spectrum encompassed BRAF V600 E and R, NRAS, the receptor tyrosine kinases FGFR2, ERBB2 as well as a passenger mutation in the protooncogene SMO (no COSMIC database entry, SIFT score 0.69). ERBB2 and FGFR2 mutations are seen in malignant melanoma, but have not yet been reported in desmoplastic malignant melanoma with the exception of a single case of an FGFR2 mutation in a desmoplastic malignant melanoma with additional sarcomatoid dedifferentiation.33 The two FGFR2 mutations seen in cases 17 and 18 are identical. This mutation (p.A259V) is a confirmed somatic mutation in metastatic renal cell carcinoma34 with a SIFT score of 0.07 and we regard it as a plausible, activating driver mutation. Finally, the ERBB2 mutation in case 14 is a previously confirmed somatic oncogenic mutation (SIFT score of 0) in breast, ovarian, and bladder carcinoma.35, 36, 37 The observed BRAF mutation frequency in mixed desmoplastic malignant melanomas at 30% is above what we expected from the literature38 with Miller et al23 reporting a 6% BRAF mutation rate in mixed desmoplastic malignant melanoma (but an absence of BRAF mutations in pure desmoplastic malignant melanoma in accordance with our data). The higher percentage of BRAF mutations in the present study in mixed desmoplastic malignant melanoma might be explained by an increased sensitivity due to manual microdissection of tumors. Desmoplastic tumor areas usually present with decreased cellularity owing to intermixed fibrosis as illustrated by one BRAF V600E mutations at a frequency at 8% of alleles (Table 2), which most likely would have been missed without microdissection. Regarding RET codon 691 status, 38% of mixed desmoplastic malignant melanomas were RETp and interestingly all three also displayed concomitant canonical activating mutations in BRAF and ERBB2.
Tumor suppressor gene mutations were found in five mixed desmoplastic malignant melanomas, namely in TP53, CDKN2A, and two tumor suppressor genes not yet reported in desmoplastic malignant melanoma: SMARCB1 and PTPN11. The mutation in SMARCB1 (case 19) is a confirmed somatic mutation in malignant melanoma39 with a SIFT score of 0, clearly arguing for biological relevance. SMARCB1 is a highly conserved subunit of the SWI/SNF ATP-dependent chromatin-remodeling complex. Its functions include facilitation of UV-induced DNA damage repair and regulatory activity of transcription and cell cycle progression.40 In accordance with its tumor suppressor function immunohistochemical loss of SMARCB1 expression has been reported as an independent prognostic factor of reduced survival in malignant melanoma.41 Lastly, the PTPN11 mutation in case 18 is an unequivocal, confirmed somatic mutation (SIFT score 0.04) previously observed in a melanoma42 and in acute lymphoblastic leukemia.43
Of note the mutation panel employed in our study also included hotspot mutation analysis for GNAQ and GNA11 two G-protein subunits leading to constitutive activation of the ERK signalling cascade. Either of these two G-protein subunits was found to be mutated in over 80% of uveal melanomas, which are almost exclusively BRAF and NRAS wild type.18, 44, 45 To the best of our knowledge this is the first study to extend GNAQ and GNA11 mutation analysis to desmoplastic malignant melanoma and our data provide evidence that tumorigenesis in desmoplastic malignant melanoma (whether of pure or mixed form) is not driven by GNAQ and GNA11 mutations.
In addition, we explored mutational heterogeneity in mixed desmoplastic malignant melanomas by separate analysis of desmoplastic and nondesmoplastic tumor areas. In four mixed desmoplastic malignant melanomas these areas were sufficiently separated to allow for selective manual microdissection. In all four cases identical mutation patterns were seen in both components, and RET polymorphism at codon 691 was identical as well.
Clinically, desmoplastic malignant melanoma as a whole is associated with a favourable prognosis, and a significantly reduced rate of lymph node metastases and this prognostic advantage has specifically been linked to the pure form. Safaee Ardekani et al46 have demonstrated a 1.7-fold reduction of overall survival in patients suffering from BRAF-mutated melanomas in their comprehensive meta-analysis. This clinical observation correlates well with the molecular findings of our study demonstrating activating mutations in the vast majority of mixed desmoplastic malignant melanomas but none in pure desmoplastic malignant melanoma. The activating mutations detected essentially all converge on the same pathways, namely activation of the MAP-kinase pathway (BRAF and RET), and additional induction of the PIK3/Akt pathway in the case of FGFR2, ERBB2, and RET. Furthermore, our study provides evidence that mixed desmoplastic malignant melanomas are homogeneous regarding their mutation profile and RETp status. RETp was detected at about the same frequency as in the general healthy population but may explain the proliferative stimulus in a subset (25% in our study) of pure desmoplastic malignant melanomas devoid of canonical oncogenic mutations and may add to or modify tumor proliferation and neurotropism in tumors with already a baseline oncogenic drive, as seen in a subset of our mixed desmoplastic malignant melanomas.
Our study thus underscores the validity of subtyping desmoplastic malignant melanoma into pure and mixed forms from a molecular perspective. Lack of canonical oncogenes in pure desmoplastic malignant melanoma might in principle be attributable to inherent limitations of a restricted hotspot mutation analysis. Specifically, complex mutations of potential relevance such as activating translocations of the anaplastic lymphoma kinase gene (ALK) have recently been described in KIT- and BRAF-negative mucosal melanomas.47 Alternatively, oncogenes amplified by copy number changes (eg, CCND1 and MYC1) were shown to exhibit site-specific frequencies in malignant melanoma.48 Regarding clinical management the dichotomy between pure and mixed desmoplastic malignant melanoma further extends to prognosis as previously reported and limits therapy options in pure desmoplastic malignant melanoma. In conclusion, especially pure desmoplastic malignant melanoma appears to be the desmoplastic malignant melanoma subentity in need for further investigation regarding additional molecular driver mechanism and structural as well as numeric genome analysis to elucidate the oncogenic changes propagating tumor growth in this special melanoma subtype.
References
Conley J, Lattes R, Orr W . Desmoplastic malignant melanoma (a rare variant of spindle cell melanoma). Cancer 1971;28:914–936.
McCarthy SW, Scolyer RA, Palmer AA . Desmoplastic melanoma: a diagnostic trap for the unwary. Pathology 2004;36:445–451.
Busam KJ . Cutaneous desmoplastic melanoma. Adv Anat Pathol 2005;12:92–102.
Chen JY, Hruby G, Scolyer RA et al. Desmoplastic neurotropic melanoma: a clinicopathologic analysis of 128 cases. Cancer 2008;113:2770–2778.
Jaroszewski DE, Pockaj BA, DiCaudo DJ et al. The clinical behavior of desmoplastic melanoma. Am J Surg 2001;182:590–595.
George E, McClain SE, Slingluff CL et al. Subclassification of desmoplastic melanoma: pure and mixed variants have significantly different capacities for lymph node metastasis. J Cutan Pathol 2009;36:425–432.
Pawlik TM, Ross MI, Prieto VG et al. Assessment of the role of sentinel lymph node biopsy for primary cutaneous desmoplastic melanoma. Cancer 2006;106:900–906.
Chapman PB, Hauschild A, Robert C et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011;364:2507–2516.
Le K, Blomain ES, Rodeck U et al. Selective RAF inhibitor impairs ERK1/2 phosphorylation and growth in mutant NRAS, vemurafenib-resistant melanoma cells. Pigment Cell Melanoma Res 2013;26:509–517.
Curtin JA, Busam K, Pinkel D et al. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 2006;24:4340–4346.
Fullen DR, Poynter JN, Lowe L et al. BRAF and NRAS mutations in spitzoid melanocytic lesions. Mod Pathol 2006;19:1324–1332.
Ellerhorst JA, Greene VR, Ekmekcioglu S et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin Cancer Res 2011;17:229–235.
Sasaki Y, Niu C, Makino R et al. BRAF point mutations in primary melanoma show different prevalences by subtype. J Invest Dermatol 2004;123:177–183.
Goel VK, Lazar AJ, Warneke CL et al. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J Invest Dermatol 2006;126:154–160.
Saldanha G, Potter L, Daforno P et al. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin Cancer Res 2006;12:4499–4505.
Wong CW, Fan YS, Chan TL et al. BRAF and NRAS mutations are uncommon in melanomas arising in diverse internal organs. J Clin Pathol 2005;58:640–644.
Edmunds SC, Cree IA, Di Nicolantonio F et al. Absence of BRAF gene mutations in uveal melanomas in contrast to cutaneous melanomas. Br J Cancer 2003;88:1403–1405.
Van Raamsdonk CD, Griewank KG, Crosby MB et al. Mutations in GNA11 in uveal melanoma. N Engl J Med 2010;363:2191–2199.
Griewank KG, van de Nes J, Schilling B et al. Genetic and clinico-pathologic analysis of metastatic uveal melanoma. Mod Pathol 2014;27:175–183.
Edwards RH, Ward MR, Wu H et al. Absence of BRAF mutations in UV-protected mucosal melanomas. J Med Genet 2004;41:270–272.
Davison JM, Rosenbaum E, Barrett TL et al. Absence of V599E BRAF mutations in desmoplastic melanomas. Cancer 2005;103:788–792.
Coupelon S, Franck F, Jarrousse AS et al. Desmoplastic malignant melanoma: a study of ten cases and status of BRAF mutation. Dermatology 2012;225:168–171.
Miller DD, Emley A, Yang S et al. Mixed versus pure variants of desmoplastic melanoma: a genetic and immunohistochemical appraisal. Mod Pathol 2012;25:505–515.
Kim J, Lazar AJ, Davies MA et al. BRAF, NRAS and KIT sequencing analysis of spindle cell melanoma. J Cutan Pathol 2012;39:821–825.
Narita N, Tanemura A, Murali R et al. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene 2009;28:3058–3068.
Takahashi M . The GDNF/RET signaling pathway and human diseases. Cytokine Growth Factor Rev 2001;12:361–373.
Sawai H, Okada Y, Kazanjian K et al. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res 2005;65:11536–11544.
Barr J, Amato CM, Robinson SE et al. The RET G691S polymorphism is a germline variant in desmoplastic malignant melanoma. Melanoma Res 2012;22:92–95.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38:e164.
Cingolani P, Platts A, Wang le L et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081.
Forbes SA, Beare D, Gunasekaran P et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811.
Kiuru M, McDermott G, Berger M et al. Desmoplastic melanoma with sarcomatoid dedifferentiation. Am J Surg Pathol 2014;38:864–870.
Rosenberg JE, Bambury RM, Van Allen EM et al. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Invest New Drugs 2014;32:178–187.
Kan Z, Jaiswal BS, Stinson J et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010;466:869–873.
Shah SP, Morin RD, Khattra J et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009;461:809–813.
Guo G, Sun X, Chen C et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet 2013;45:1459–1463.
Greaves WO, Verma S, Patel KP et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn 2013;15:220–226.
Wei X, Walia V, Lin JC et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet 2011;43:442–446.
Vries RG, Bezrookove V, Zuijderduijn LM et al. Cancer-associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes Dev 2005;19:665–670.
Lin H, Wong RP, Martinka M et al. Loss of SNF5 expression correlates with poor patient survival in melanoma. Clin Cancer Res 2009;15:6404–6411.
Krauthammer M, Kong Y, Ha BH et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 2012;44:1006–1014.
Tartaglia M, Martinelli S, Cazzaniga G et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood 2004;104:307–313.
Van Raamsdonk CD, Bezrookove V, Green G et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009;457:599–602.
Populo H, Vinagre J, Lopes JM et al. Analysis of GNAQ mutations, proliferation and MAPK pathway activation in uveal melanomas. Br J Ophthalmol 2011;95:715–719.
Safaee Ardekani G, Jafarnejad SM, Tan L et al. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PLoS One 2012;7:e47054.
Niu HT, Zhou QM, Wang F et al. Identification of anaplastic lymphoma kinase break points and oncogenic mutation profiles in acral/mucosal melanomas. Pigment Cell Melanoma Res 2013;26:646–653.
Glatz-Krieger K, Pache M, Tapia C et al. Anatomic site-specific patterns of gene copy number gains in skin, mucosal, and uveal melanomas detected by fluorescence in situ hybridization. Virchows Arch 2006;449:328–333.
Acknowledgements
The used biospecimens for this research project were provided by Biobank Graz. The authors thank the following pathologists for providing material of previous consultation cases for this study: Dr M Hackl, Landesklinikum Amstetten, Austria; Dr R Stering, Landesklinikum Leoben, Austria; and Dr R Tarmann, Landesklinikum Klagenfurt, Austria.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Jahn, S., Kashofer, K., Halbwedl, I. et al. Mutational dichotomy in desmoplastic malignant melanoma corroborated by multigene panel analysis. Mod Pathol 28, 895–903 (2015). https://doi.org/10.1038/modpathol.2015.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.39
