
Abstract
TEMPI syndrome (telangiectasias, elevated erythropoietin level and erythrocytosis, monoclonal gammopathy, perinephric fluid collections, and intrapulmonary shunting) is a recently described syndrome that, owing to erythrocytosis, may be confused with polycythemia vera. It is best classified as a type of plasma cell dyscrasia with paraneoplastic manifestations, similar to POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin abnormalities). To date, 11 patients have been identified. This is the first morphologic review of TEMPI syndrome bone marrow samples, in order to define pathologic features that may aid in the recognition of the syndrome and to identify post-therapy changes. Seven bone marrow aspirates and biopsies from three patients, including two post-treatment marrows, were examined. Patients were 36, 49, and 49 years old at time of diagnosis. In all cases, erythropoietin levels were extremely elevated at >5000 IU/l, the paraprotein was IgG kappa, JAK2 V617F was negative and vascular endothelial growth factor levels were normal. In one case, the increase in clonal plasma cells reached levels of smoldering myeloma (18%), but remaining marrows showed few monoclonal plasma cells (<5%). All pre-treatment biopsies showed erythroid hyperplasia, with mild nonspecific megakaryocytic, and erythroid cytologic atypia in one marrow. Prominent plasma cell vacuolization and reactive-appearing lymphoid aggregates were noted in one case. Findings of myeloproliferative neoplasms, including megakaryocyte clusters and fibrosis, were not identified. In conclusion, TEMPI syndrome should be considered when erythrocytosis and plasma cell dyscrasia coexist. The bone marrow findings, although nonspecific, differ significantly from polycythemia vera. Peculiar clinical and laboratorial findings of TEMPI, including elevated erythropoietin and normal vascular endothelial growth factor level, allow the diagnosis and distinction from POEMS syndrome. Significant decrease in erythropoietin level following treatment suggests a role of erythropoietin in monitoring therapeutic response.
Similar content being viewed by others
Main
TEMPI syndrome is a plasma cell dyscrasia with paraneoplastic manifestations described in 2011 by Sykes et al.1 It is defined by T—denoting telangiectasias, E—denoting erythrocytosis with elevated erythropoietin levels, M—denoting monoclonal gammopathy, P—denoting perinephric fluid collections, and I— denoting intrapulmonary shunting. To our knowledge, 11 patients have been identified.1, 2, 3, 4, 5, 6 The mechanisms of disease and epidemiologic findings, including prevalence, are unknown. Significant clinical improvement has been observed using a bortezomib-based regimen,2, 6 and in one case followed by autologous stem cell transplant.7 This therapy is similar to that recommended for other plasma cell neoplasms.
Among the 11 patients reported, 8 underwent at least one bone marrow biopsy prior to the diagnosis of TEMPI. Indication for the procedure was to either exclude polycythemia vera in the setting of unexplained erythrocytosis or to determine plasma cell burden following abnormal serum protein electrophoresis. Bone marrow morphologic findings of TEMPI, at diagnosis and post therapy, have not been reported in detail.
This is the first detailed morphologic description of histopathologic and clinical findings of a series of TEMPI syndrome cases. This study aims to define histopathologic features that may aid in the diagnosis of future cases, and to identify post-therapy changes.
Materials and methods
We obtained seven archival bone marrow specimens from three patients, including two post-treatment samples. Materials were obtained from the institutions at which the diagnosis of TEMPI syndrome was originally made. Institutional review board approval for this study was sought and obtained from each contributing institution. The material comprised Wright–Giemsa-stained aspirate smears, bone marrow trephine hematoxylin and eosin–stained core biopsies, Prussian blue iron, and reticulin special stains. The slides were independently reviewed by at least two hematopathologists. Specific pre- and post-treatment parameters were assessed as follows: overall bone marrow cellularity; quantities and morphologic features of erythrocytes, granulocytes, megakaryocytes, plasma cells, and lymphocytes; presence of fibrosis; and abnormalities in bone trabeculae and sinuses. Clinical and demographic information and relevant radiological and laboratorial data were obtained from collaborating pathologists and/or oncologists, including complete blood count values at time of biopsy, type of serum paraprotein, serum erythropoietin levels, and other pertinent ancillary studies including flow cytometry and molecular genetic studies. Clinical findings of two patients have been previously reported.1, 6
Results
Patients (designated A, B, and C) were 36, 49, and 49 years old, respectively, at the time of diagnosis, and comprised two females and one male (Table 1). Erythrocytosis was initially present in all patients. However, at time of diagnosis, patients A and C had developed iron deficiency anemia while being phlebotomized for a presumed diagnosis of polycythemia vera. At time of diagnosis, erythropoietin ranged from 4160 to 8144 mU/ml (normal Epo: 2.6–18.5 mU/ml). Following chemotherapy in patient C, erythropoietin level decreased to 1793 mU/ml and was 95.4 mU/ml after autologous stem cell transplant. All patients had IgG kappa light chain-restricted plasma cells, identified by either immunofixation and/or flow cytometry. Perinephric fluid collections were identified incidentally in all patients. Abnormal intrapulmonary right-to-left intravenous shunting was detected by lung perfusion studies. All patients developed venous thrombosis as a complication, one of them while in clinical disease remission (patient C).
Data from pertinent ancillary studies were obtained. JAK2 V617F molecular genetic testing was negative in all cases. Molecular genetic studies for hereditary hemorrhagic telangiectasia and hemoglobin oxygen affinity studies were negative in two patients. Chromosome analysis performed on bone marrow samples, including myeloma fluorescent in situ hybridization panels, were normal in all cases. Additional negative studies performed in all patients included hemoglobin electrophoresis, beta globin gene sequencing, serum vascular endothelial growth factor levels, and von Hippel-Lindau gene mutation analysis.
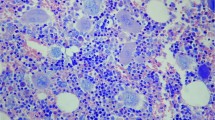
Bone marrow morphological findings are summarized in Table 2. Erythroid hyperplasia in all patients, combined with megakaryocytic hyperplasia in patients A and C, accounted for the increase in bone marrow cellularity noted in all cases. No morphologic features of chronic myeloproliferative neoplasms were identified. Specifically, none of the cases showed characteristic megakaryocytic hyperchromia or clustering, increased reticulin fibrosis, osteosclerosis, or intrasinusoidal hematopoiesis that can be seen in long standing polycythemia vera or other myeloproliferative neoplasms. In patient A, the increase in clonal plasma cells (18%) reached the level of smoldering myeloma, whereas in patients B and C, plasma cells were at levels typically seen in monoclonal gammopathy of undetermined significance (<5%). Slight plasma cell atypia was noted in all cases, with prominent vacuolization in patient A (see Figure 1). Erythroid hyperplasia and moderate atypia (frequent binucleation and nuclear blebbing), as well as mild megakaryocytic atypia (occasional monolobated forms), were seen in two of three pre-treatment samples (Figure 1). Mild erythroid atypia persisted in post-treatment samples. Non-paratrabecular lymphoid aggregates were noted in pre and post-treatment samples (patients B and C). These contained small normal-appearing lymphocytes without conspicuous association with any other cell types, namely plasma cells or mast cells.

Morphologic findings in TEMPI syndrome. Plasma cell clusters showed prominent cytoplasmic vacuolization in patient A (a), and frayed cytoplasm in patient B (b). All cases showed erythroid hyperplasia and atypia, predominantly nuclear blebbing (c), in the setting of superimposed iron deficiency anemia. Features of chronic myeloproliferative neoplasms are not present, specifically, no megakaryocyte clustering or atypia (d).
Significant clinical, laboratorial, and morphologic improvement was observed following treatment in the single patient treated with bortezomib and stem cell transplant (patient C) included in this series. The patient’s severe hypoxemia resolved, erythropoietin levels decreased, and bone marrow plasma cell clones or monoclonal protein by serum protein electrophoresis became undetectable.
Discussion
TEMPI syndrome is a newly described type of plasma cell dyscrasia of unknown prevalence, pathogenesis, and prognosis. To date, 11 patients have been identified.1, 2, 3, 4, 5, 6 During the course of preparation of this manuscript, an additional patient was reported,5 thus totaling five new patients since the initial six cases described in 2011.1 It is certain that more patients with undiagnosed TEMPI syndrome exist and would benefit from a specific diagnosis, as symptomatic improvement may be achieved with a bortezomib-based regimen.2, 6 In an effort to better characterize this disease and improve its recognition among pathologists and clinicians, we undertook a detailed analysis of serial bone marrow findings in patients with known TEMPI syndrome.
Our study shows that in these few cases, there are no specific morphologic findings of TEMPI syndrome within the bone marrow. Unremarkable bone marrow evaluations have also been described in other cases of TEMPI syndrome.2, 3, 4 Slight atypia noted in erythroid and megakaryocytes could be attributed to either superimposed iron deficiency anemia or to treatment effect. The pathologic findings, albeit nonspecific, significantly differ from chronic myeloproliferative neoplasms such as polycythemia vera. Prominent plasma cell vacuolization was noted in one case (Patient A, Figure 1 and Table 2). This type of plasma cell vacuolization has been previously described in glycogenosis8 and μ-heavy chain disease.9 The significance of this finding in TEMPI syndrome is unknown.
TEMPI syndrome is best classified as a plasma cell dyscrasia with accompanying paraneoplastic manifestations, similar to POEMS and Schnitzler syndromes. Patients with POEMS syndrome (defined by polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin abnormalities) may also present with erythrocytosis,10 raising the differential diagnosis of TEMPI syndrome in certain circumstances. In contrast to POEMS syndrome, in which vascular endothelial growth factor is elevated,10 no increase in vascular endothelial growth factor was seen in cases described in this study and other cases of TEMPI syndrome.4 Schnitzler syndrome is a rare disorder characterized by monoclonal gammopathy, urticarial rash, fever, arthritis, hepatosplenomegaly, lymphadenopathy, and leukocytosis. In contrast to TEMPI and POEMS syndromes, patients can develop anemia of chronic inflammation, rather than erythrocytosis.10, 11 Although identification of erythrocytosis can be valuable in the differential diagnosis of these three entities, concurrent iron deficiency in TEMPI syndrome masked the finding of polycythemia in the cases described in this series. Morphologically, the lymphoid aggregates noted in this study of TEMPI were not rimmed by plasma cells, in contrast with those aggregates seen in POEMS.12 In Schnitzler syndrome, bone marrow examinations are either normal or show polytypic lymphocytic or plasmacytic infiltrates.13 The clinical features of TEMPI, POEMS, and Schnitzler syndromes are illustrated in Table 3.
The monoclonal paraprotein was identified as IgG kappa in this study and in six of eight cases of TEMPI syndrome.1, 5, 7 IgG lambda and IgA lambda have been reported in one patient each.2, 4 In POEMS syndrome, lambda light chain predominates, and in Schnitzler syndrome, almost all patients have an IgM kappa paraprotein.12 In this study, plasma cell burden reached 18% in patient A, and up to 15% in one of the reported cases.2 The other two cases (patients B and C) had plasma cell percentages at levels of monoclonal gammopathy of undetermined significance (<10%), similar to POEMS and Schnitzler syndromes,13, 14, 15 and other previously reported TEMPI cases.1, 3, 4, 5 Although renal dysfunction has been described in one patient,4 none of the patients with TEMPI syndrome fulfill criteria for plasma cell myeloma.1, 2, 3, 5, 6
Erythropoietin levels are helpful in the differential diagnosis of TEMPI syndrome and other causes of erythrocytosis. Erythropoietin levels in TEMPI ranged from 78 mU/ml to 8144 mU/ml (median 500 mU/ml) at presentation in 9 of 11 patients.1, 2, 3, 4, 5, 6, 7 Such elevated values are not seen in most erythropoietin-driven erythrocytoses, although levels as high as 3000 mU/ml have been noted in patients with VHL mutations.16 Secondary erythrocytosis (owing to hypoxemia or procedure such as phlebotomy) typically elevates erythropoietin levels to no greater than 30 mU/ml.17 In polycythemia vera or other primary erythrocytoses, erythropoietin is low (average <3.3 mU/ml). In TEMPI, erythropoietin levels are highest in patients with severe oxygen dependent hypoxemia (in our study, patients B and C) and at ∼100 mU/ml in patients with mild hypoxemia not requiring oxygen therapy.2, 3
Erythrocytosis appears to be the initial clinical manifestation of this disease, followed by the slow progression to severe and debilitating hypoxemia.1, 6 Hypoxemia has been attributed to right–left intrapulmonary shunting, although in our study, associated iatrogenic iron deficiency anemia secondary to therapeutic phlebotomy for a presumed diagnosis of polycythemia vera may have contributed to this symptom. In this study, the clinical course of all patients was complicated by venous thrombosis, which was also previously reported in an additional patient.1 It has been suggested that the perinephric collections of serous fluid seen in TEMPI syndrome are related to malformation of lymphatic vessels.4 Therapy directed against the plasma cell clone has yielded excellent clinical results, with resolution of hypoxemia, normalization of red cell counts, and regression of telangiectasias in three patients treated, as previously reported.2, 6, 7 Morphologically, the single patient treated in this study (patient C) had normalization of bone marrow cellularity owing to resolution of erythroid hyperplasia (Table 2). The morphologic response is likely, at least in part, due to correction of iatrogenic iron deficiency anemia with discontinuation of phlebotomy. Mild erythroid atypia (predominantly nuclear blebbing) persisted in post-therapy samples in patient C, which may have been due to treatment effect. In addition, erythropoietin levels decreased and plasma cell clonality became undetectable by high-sensitivity flow cytometry. In previously reported cases, erythropoietin levels also decreased significantly after initiation of therapy.2, 6 This may suggest a role of erythropoietin as a potential marker of treatment response.
Although the pathogenesis of TEMPI syndrome is unknown, the remarkable response seen with bortezomib-based regimens suggests a central role of the plasma cell clone. It has been hypothesized that the monoclonal protein may trigger the paraneoplastic symptoms of TEMPI syndrome through immunologic mechanisms, and potential antigenic targets are currently under investigation.6, 7 Paraneoplastic ectopic production of erythropoietin, similar to that seen in other hematopoietic and solid malignancies,18 is an alternative hypothesis. However, increased erythropoietin alone would not explain all of the symptoms. Imaging studies in TEMPI syndrome failed to identify a source of ectopic erythropoietin production.2 Studies to elucidate disease mechanisms performed in this series and in previous reports have yielded negative or normal results.3, 4
In summary, the diagnosis of TEMPI syndrome should be considered when erythrocytosis and plasma cell dyscrasia coexist. In this setting, determination of erythropoietin levels can be helpful, as extremely elevated serum erythropoietin is a peculiar manifestation of the disease. Upon confirmation of the erythrocytosis, identification of a plasma cell clone, in conjunction with findings of clinical examination and imaging studies will allow for confirmation of the diagnosis. Normal vascular endothelial growth factor level and other clinical findings should allow the distinction from POEMS and Schnitzler syndrome. Establishing the diagnosis of TEMPI syndrome is critical, as patients who are treated appropriately have an indolent clinical course. However, patients who remain undiagnosed may ultimately develop debilitating hypoxemia. Awareness of TEMPI syndrome and its constellation of findings is important for pathologists, who may examine bone marrow biopsies from these patients at the time of presentation as part of evaluation of a monoclonal protein and/or unexplained erythrocytosis, or following treatment to assess for disease response.
References
Sykes DB, Schroyens W, O'Connell C . TEMPI Syndrome – a novel multisystem disease. N Engl J Med 2011;365:475–477.
Kwok M, Korde N, Landgren O . Bortezomib to treat the TEMPI syndrome. N Engl J Med 2012;366:1843–1845.
Mohammadi F, Wolverson MK, Bastani B . A new case of TEMPI syndrome. Clin Kidney J 2012;5:556–558.
Viglietti D, Sverzut JM, Peraldi MN . Perirenal fluid collections and monoclonal gammopathy. Nephrol Dial Transplant 2012;27:448–449.
Ryden A, Wei K, Rodriguez R et alToo Much Blood: A Case of the Newly Described TEMPI Syndrome. Chest 2013, 144:4 (meeting abstracts).
Schroyens WA, O'Connell CL, Lacy MQ et alTEMPI a reversible syndrome following treatment with bortezomib. Blood 2012; 367:778-780.
Schroyens W, O'Connell C, Sykes DB . Complete and partial responses of the TEMPI syndrome to bortezomib. N Engl J Med 2012;367:778–780.
Pralle H, Schröder R, Löffler H . New kind of cytoplasmic inclusions of plasma cells in acid maltase deficiency. Acta Haematol 1975;53:109–117.
Liapis K, Apostolidis J . Empty, but heavy, plasma cells. Blood 2012;120:4282.
Merlini GL, Palladini G . Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012;2012:595–603.
de Koning HD1, Bodar EJ, van der Meer JW et alSchnitzler Syndrome Study Group. Schnitzler syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum 2007;37:137–148.
Dao LN, Hanson CA, Dispenzieri A et al. Bone marrow histopathology in POEMS syndrome: a distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood 2011;117:6438–6444.
Lipsker D . The Schnitzler syndrome. Orphanet J Rare Dis 2010;5:38.
Jain T, Offord CP, Kyle RA et al. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica 2013;98:1581–1585.
Scarlato M, Carpo M, Previtali S et al. Poems syndrome: role of two angiogenic factors, VEGF and EPO. J Peripher Nerv Syst 2004;9:110.
Bond J, Gale DP, Connor T et al. Dysregulation of the HIF pathway due to VHL mutation causing severe erythrocytosis and pulmonary arterial hypertension. Blood 2011;117:3699–3701.
Mossuz P, Girodon F, Donnard M et al. Diagnostic value of serum erythropoietin level in patients with absolute erythrocytosis. Haematologica 2004;89:1194–1198.
Al-Tourah AJL, Tsang PW, Skinnider BF et al. Paraneoplastic erythropoietin-induced polycythemia associated with small lymphocytic lymphoma. J Clin Oncol 2006;24:2388–2389.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Rosado, F., Oliveira, J., Sohani, A. et al. Bone marrow findings of the newly described TEMPI syndrome: when erythrocytosis and plasma cell dyscrasia coexist. Mod Pathol 28, 367–372 (2015). https://doi.org/10.1038/modpathol.2014.117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.117
