
Abstract
ALK-positive large B-cell lymphoma is an aggressive lymphoid neoplasm characterized by a monomorphic proliferation of immunoblast-like cells expressing a plasmablastic phenotype and carrying ALK rearrangements. MYC rearrangements are frequent in plasmablastic lymphomas, advanced plasma cell myelomas and a subgroup of diffuse large B-cell lymphomas, but their presence in ALK-positive large B-cell lymphomas is unknown. MYC expression is downregulated by BLIMP1, a master modulator of plasma cell differentiation. BLIMP1 and MYC are upregulated by STAT3, a signal transducer activated by ALK. To determine the role of BLIMP1, MYC and STAT3 in the pathogenesis of ALK-positive large B-cell lymphomas, we investigated MYC rearrangement and the expression of MYC, phosphorylated STAT3, BLIMP1, PAX5 and XBP1 in 12 ALK-positive large B-cell lymphomas. All cases expressed ALK with a granular cytoplasmic pattern. Nine cases had a split signal consistent with an ALK rearrangement. Three additional cases showed a deletion of the 5′ or 3′ end of the ALK probe consistent with cryptic translocation. PAX5 was virtually negative in all cases tested, whereas BLIMP1 was expressed in all tumors and XBP1 in 11 of 12. Phosphorylated STAT3 was observed in all cases with a strong and diffuse nuclear pattern. MYC rearrangements were not identified in any tumor, but MYC gains and amplification were detected in six cases and one case, respectively. MYC protein was expressed in all tumors independently of MYC gene alterations. These results indicate that ALK-positive large B-cell lymphomas express a complete plasmablastic differentiation program but, contrary to plasmablastic lymphomas, do not have MYC rearrangements. STAT3 is constantly activated and may be an alternative mechanism to promote MYC expression in these tumors. The relevance of the ALK/STAT3 pathway in the pathogenesis of ALK-positive large B-cell lymphomas may offer an attractive target for new therapies.
Similar content being viewed by others
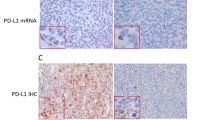

Main
ALK-positive large B-cell lymphoma was initially described by Delsol et al1 in 1997 as an uncommon and aggressive subtype of diffuse large B-cell lymphoma with frequent immunoblastic morphology, a plasma cell phenotype and expression of the ALK protein. This protein is detected in most cases with a granular cytoplasmic pattern, which is commonly associated with the rearrangement of ALK with the clathrin gene CTCL, t(2;17).2 However, other genetic partners as well as other patterns of cell localization of the ALK protein have been described.3, 4, 5 In addition to the ALK rearrangements, conventional cytogenetic studies have reported the presence of frequent complex karyotypes with multiple structural and numerical alterations in other chromosomes.2, 6, 7, 8, 9, 10, 11, 12, 13, 14
The oncogenic mechanisms involved in the pathogenesis of human ALK-positive large B-cell lymphomas are not well known. ALK protein is a cytoplasmic receptor with tyrosine kinase activity that is not normally expressed in lymphoid cells. ALK rearrangements upregulate oncogenic fusion proteins in which the ALK fragment contains the catalytic domain and the fused partner provides a dimerization domain that activates the receptor without the need of the ligand.15, 16 ALK activation has the oncogenic potential in different types of cells including B and T lymphocytes, and in transgenic mice, it induces the development of plasma cell tumors.17, 18 The dimerization of ALK leads to the activation of different downstream signaling pathways, one of the most relevant being the STAT3 pathway.19, 20 STAT3 is an important signal transducer that triggers the program of plasma cell differentiation through BLIMP1 activation, XBP1 and IRF4 expression.21 ALK-positive large B-cell lymphomas have a phenotype similar to that of plasma cells, being negative for CD20 and positive for CD138. However, whether these tumors express a complete terminal B-cell differentiation program with the upregulation of BLIMP1 and XBP1 is not well known.22 STAT3 also regulates many proteins involved in tumor cell proliferation including MYC that may have an important role in the pathogenesis of these tumors.23
The aggressive behavior of some B-cell lymphomas has been correlated with the presence of MYC rearrangement. This genetic alteration has been observed frequently in lymphoid neoplasms with plasma cell differentiation, such as plasmablastic plasma cell myelomas and plasmablastic lymphomas.24, 25, 26 On the other hand, MYC protein expression has been recently described in some aggressive B-cell lymphomas, not always related to the presence of MYC gene alterations.27, 28, 29, 30 The presence of MYC alteration and protein expression, as well as the activation of STAT3 in ALK-positive large B-cell lymphomas are not well known.
The aims of this study were to determine whether ALK-positive large B-cell lymphomas express a complete terminal B-cell differentiation program with the upregulation of BLIMP1 and XBP1 and whether MYC genetic alterations and STAT3 activation may play a role in the pathogenesis of these tumors.
Materials and methods
Case Selection
Twelve ALK-positive large B-cell lymphoma cases were retrieved from the files of the Pathology Department of the Hospital Clinic of Barcelona; Netherlands Cancer Institute (Amsterdam, The Netherlands); Laboratorio de Hematopatología (Mendoza, Argentina) and Consultoria em Patologia (Botucatu, Brazil). The patients were predominantly males (92%) with a median age of 33 years (range 22–42 years). Ten patients (83%) presented with nodal disease and two had extranodal presentation: one in the right colon and the other one in the soft tissues of the thigh (Table 1).
For comparison purposes, we also studied 11 plasmablastic lymphomas and 16 diffuse large B-cell lymphomas that were selected because they were of the activated B-cell subtype by gene expression profiling (11 cases) or had been classified immunohistochemically as non-germinal center-like subtype using the Hans’ algorithm (5 cases).31
Immunohistochemistry
Immunohistochemical studies were carried out with a panel of monoclonal and polyclonal antibodies reactive in paraffin-embedded tissue sections using a peroxidase-labeled detection system, standard antigen retrieval protocols and an automated immunostainer (Dako Autostainer, Glostrup, Denmark and Bond-Max, Leica Microsystems, Wetzlar, Germany). The panel of antibodies used included CD20 (clone L26; Dako), CD79a (clone JCB117; Dako), PAX5 (clone 24; BD Biosciences), CD30 (clone Ber-H2; Dako) and EMA (clone E29; Dako). Polyclonal antibodies for IgA, IgG, kappa and lambda were applied (all from Dako). ALK expression was determined using the monoclonal ALK1 antibody (Dako).The terminal B-cell differentiation program was evaluated using antibodies anti-IRF4 (clone MUM1p; Dako) and CD138 (clone MI15; Dako), PRDM1/Blimp1 (clone Sc13206; Santa Cruz Biotechnology) and XBP1 (clone Sc7160; Santa Cruz Biotechnology). The conditions for all these antibodies were the same as described previously.32 A cutoff of 25% was used to interpret the results as positive.
MYC expression was assessed using the monoclonal Y69 antibody (Epitomics), at dilution 1/40, with incubation of the primary antibody for 1 h after antigen retrieval at pH 6 for 30 min. The activation of STAT3 was analyzed using the monoclonal phosphorylated-STAT3 (pSTAT3, phosphorylation on tyrosine 705) antibody (clone D3A7; Cell Signaling Technology). The samples were incubated with this antibody at a dilution of 1:100 for 2 h at 37 °C. MYC and pSTAT3 immunostaining were evaluated in a semiquantitative manner and a normal tonsil tissue was used as the control. A cutoff of 25% was also used to interpret the results as positive.
The presence of Epstein–Barr virus was examined by in situ hybridization to detect Epstein–Barr virus-encoded early nuclear RNAs. The latency-associated nuclear antigen 1 of human herpes virus type 8 was studied by immunohistochemistry using the clone LN53 (Advanced Biotechnologies), both as described earlier.25
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization was performed on 3- to 4-μm-thick sections of formalin-fixed, paraffin-embedded tissues, using break-apart probes (Vysis, Abbott Molecular) specific for ALK (2p13) and MYC (8q24) as described previously.25
Tonsil sections were used as controls. For every tumor and tonsil sample, a minimum of 100 evaluable nuclei were scored. The cutoff values for the interphase FISH analyses were established following the criteria of Ventura et al.33 To detect rearrangements, the cutoff value was 3%. Moreover, the mean number of numerical genetic alterations was evaluated in each case to assess the incidence of the genetic events in the tumors. Gains were considered when three to four copies of the gene studied were identified, whereas more than four copies were considered as amplification. A deletion was considered when one of the alleles lost one of their extremes in >40% of the neoplastic cells.
Results
Pathological Features of ALK-Positive Large B-Cell Lymphomas
The clinical and immunophenotypic features of the 12 ALK-positive large B-cell lymphomas are summarized in Table 1. All cases had a granular cytoplasmic distribution of ALK expression. CD20 was negative in all of them. CD79a, PAX5 and CD30 were detected in isolated cells in 2/9, 1/10 and 2/12 cases, respectively, but none of the tumors had positivity for more than one of these B-cell markers. EMA was expressed in all cases. IgA was positive in 9/12 cases and two of them also expressed IgG. No clear expression of immunoglobulin heavy chains could be demonstrated in three cases. Lambda restriction was detected in 7/12 and kappa restriction in 2/12, and in 3 the study was not contributory. Epstein–Barr virus and/or human herpes virus type 8 were negative in all the cases investigated.
CD138 was positive in all cases examined. IRF4 was positive in 8/11 (73%) cases; two had weak expression in 10–25% of the neoplastic cells and one was negative. BLIMP1 was positive in all cases, most of them with strong expression in >50% of the cells. Similarly, XBP1 was also expressed in 11 of 12, although the number of positive cells was slightly lower than for BLIMP1.
The ALK break-apart probe showed different patterns of rearrangement and alterations in all cases (Table 1 and Figure 1). Seven of 12 (58%) cases showed a split ALK signal consistent with a chromosomal rearrangement (cases 1, 3, 4, 5, 7, 10 and 12). Two of these cases also had extra normal copies of the normal allele (cases 3 and 4). A split signal of both alleles was observed in two other cases (cases 2 and 8) and one of them also showed extra copy signals of normal ALK allele (case 8). Two other cases exhibited isolated copies of 3′ end (telomeric region) (cases 9 and 11), one of them with additional copies of the normal ALK (case 11). One additional case showed isolated extra copies of the 5′ end of ALK (centromeric region) and of the normal allele (case 6). These three cases with deletions of the 3′ or 5′ ends had ALK protein expression with the same granular cytoplasmic pattern as the other cases.

Different ALK patterns detected by fluorescence in situ hybridization (FISH). (a) Normal ALK pattern observed in a reactive lymph node; (b) case 1, simple rearrangement; (c) case 3, simple rearrangement and extra copies; (d) case 8, double rearrangement; (e) case 2, double rearrangement and extra copies; (f) case 6, three loci with deletion of the 3′ end and extra copies; (g) case 9, deletion of the 5′ end; (h) case 11, three loci with deletion of the 5′ end and extra copies. Yellow arrow: normal signal of ALK; red arrow: split signal of ALK, 3′ end; and green arrow: split signal of ALK, 5′ end.
MYC Gene Alterations and Protein Expression
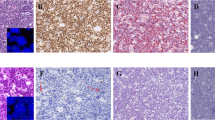
The genetic alterations and protein expression of MYC in ALK-positive large B-cell lymphomas, plasmablastic lymphomas and diffuse large B-cell lymphomas are summarized in Table 2 and Figure 2. MYC analysis by FISH was successful in the 11 ALK-positive large B-cell lymphomas cases studied. Six cases showed an increased copy number of the gene, 5 gains (cases 4, 5, 10, 11 and 12) and 1 amplification (case 3), whereas 5 cases did not show any genetic alteration. The mean copy number gains per cell was 3.26 (range 2.76–3.87) and the amplified copies 5.84. No evidence of MYC rearrangement was detected in any case.

Phosphorylated signal transducers and activators of transcription-3 (pSTAT3) and MYC in ALK+ large B-cell lymphoma (LBCL), plasmablastic lymphoma (PBL) and activated B-cell-like (ABC) diffuse large B-cell lymphoma (DLBCL). (a) Case 3, MYC amplified in an ALK+ LBCL; (b) case 3, MYC protein expression in >75% of the cells; (c) case 3, pSTAT3 expression in all tumor cells; (d) case 13, MYC rearranged in a PBL case; (e) case 13, MYC protein expression in 50–75% of the neoplastic cells; (f) case 13, pSTAT3 protein expression in 50–75% of the tumor cells. Immunohistochemistry (IHC) images (b, c, e and f) photographed at × 400. Yellow arrow: normal signal of MYC; red arrow: split signal of MYC, 5′ end; green signal: split signal of MYC, 3′ end.
MYC protein was expressed in the 12 cases. Six tumors had MYC positivity in 25–50% of the cells and three of them had MYC gains (cases 10, 11 and 12). Three cases had MYC expression in 50–75% of the neoplastic cells, and two of them had MYC gains (cases 4 and 5). The other three tumors showed MYC expression in >75% of the tumor cells and one of them demonstrated amplification of the gene (case 3). No gene alterations were observed in six cases.
In plasmablastic lymphomas, MYC genetic alterations were observed in 9/11 cases, 4 rearrangements and 5 gains. MYC expression was detected in 7/11 cases. Four cases were positive in 25–50% of the neoplastic cells, two of them showed MYC gains (cases 16 and 19), one had a gene rearrangement and no gene alterations were observed in one. Three tumors were MYC positive in 50–75% and all of them had MYC rearrangement (cases 13, 14 and 18). No MYC expression was observed in three plasmablastic lymphomas with MYC gains (cases 17, 20 and 21) (Table 2).
In diffuse large B-cell lymphomas, MYC genetic alterations were observed in 2/16 cases, one rearrangement and one gain. MYC expression was detected in 7/16 cases. Two cases showed MYC expression in >75% of the neoplastic cells, both without any MYC gene alteration (cases 24 and 35). In two cases, MYC expression was observed in 50–75% of the tumor cells, but none of them showed MYC gene alterations by fluorescence in situ hybridization (cases 36 and 37). Three additional cases showed MYC expression in 25–50% of the neoplastic cells (cases 25, 26 and 27) and one of them presented rearrangement of the gene (case 25).
pSTAT3 Expression
pSTAT3 expression in ALK-positive large B-cell lymphomas, plasmablastic and diffuse large B-cell lymphoma-activated B-cell subtype is detailed in Table 2 and Figure 2. All ALK-positive large B-cell lymphomas were strongly positive for pSTAT3. This overexpression was observed in virtually all neoplastic cells, except in one case (case 12).
In plasmablastic lymphomas, pSTAT3 expression was observed in 4/6 cases. Three of them (cases 13, 15 and 19) showed pSTAT3 expression in 50–75% of the neoplastic cells. MYC expression correlated with pSTAT3 expression in these cases. The fourth case (case 21) showed pSTAT3 expression in virtually 100% of the neoplastic cells, but MYC expression was negative. Two cases were negative for pSTAT3, one expressed MYC in 25–50% and the other was negative.
pSTAT3 expression was detected in 4/11 diffuse large B-cell lymphoma cases in 50–75% of the neoplastic cells. Two of these cases (cases 24 and 37) also showed MYC expression in a similar percentage, whereas the other two cases were negative for MYC (cases 28 and 38). None of them had MYC genetic alterations. Three cases without pSTAT3 expression (cases 27, 35 and 36) expressed MYC, but none of them showed genetic alterations of the gene.
Discussion
ALK-positive large B-cell lymphoma is an uncommon subtype of diffuse large B-cell lymphoma carrying ALK rearrangement and protein expression. The most common gene partner of ALK in these tumors is CLTC at 17q232, 8, 14, 34, 35, 36, 37, 38, 39, 40, 41 followed by NPM1 at 5q356, 10 and other genes identified in occasional cases such as SQSTM142 and SEC31A.13 Genetic studies have recognized other translocations between ALK and chromosomal regions at 4q22, 12q24 or Xq21, but the corresponding gene partner has not been identified.11, 12 All these translocations lead to the overexpression and oncogenic activation of the ALK protein. The CLTC-ALK fusion generates a protein recognizable by immunohistochemistry with a cytoplasmic granular pattern. However, this pattern is also detected in other translocations, with the exception of the nuclear and cytoplasmic pattern of the NPM1-ALK fusion. All our cases had ALK expression with a cytoplasmic granular pattern, although the partner involved could not be determined since we used a break-apart ALK probe. A split signal consistent with an ALK translocation was detected in 7 of the 12 cases. Intriguingly, two cases showed only a deletion of the 5′ end of the probe. This FISH pattern has been observed occasionally in ALK-positive large B-cell lymphomas.4, 11, 12, 13 The combination of FISH and molecular studies have confirmed that this pattern is due to cryptic translocations of the 3′ end of ALK fused with different partners and associated with the deletion of the ALK 5′ end. Similar to the reported cases, our two tumors with the 5′ end deletion expressed ALK with the same granular cytoplasmic pattern as the other cases with the typical split signal. In addition to the rearranged gene, our FISH study detected double splits in two cases and extra copies of the normal ALK gene in five cases. These findings are consistent with the multiple chromosomal alterations and karyotypes close to the tetraploidy detected in around 50% of these tumors by conventional cytogenetics.2, 6, 7, 8, 9, 10, 11, 12, 13, 14 However, as we have not used centromer probe in this study, we cannot distinguish between true polysomies or partial chromosomal gains. We also found a case with deletion of 3′ end, which has not been reported before in the literature. This case also expressed ALK protein with the same granular and cytoplasmic pattern of expression, suggesting that ALK may have a cryptic rearrangement with a similar functional effect upregulating ALK protein.
The oncogenic pathways involved in the pathogenesis of ALK-positive large B-cell lymphomas are not well defined. ALK rearrangements produce fusion proteins with constitutive tyrosine kinase activity, which activates several downstream pathways. One of them is the activation of STAT3 signaling that seems to be required for ALK-mediated lymphomagenesis in different cell models including murine and human B and T cells.19, 20, 43 In this study, we have demonstrated that activated pSTAT3 is strongly expressed in all ALK-positive large B-cell lymphomas. These observations expand the previous identification of pSTAT3 in two ALK-positive large B-cell lymphomas44 and suggest that this pathway may be important in the pathogenesis of these lymphomas. The relevance of STAT3 in ALK-positive large B-cell lymphomas is highlighted by its downregulation in cell lines derived from these tumors treated with an ALK inhibitor.7, 13
The strong and constant expression of pSTAT3 in ALK-positive large B-cell lymphomas contrasted with the lower expression observed in non-germinal center B-cell-like diffuse large B-cell lymphomas (36%) and slightly lower in plasmablastic lymphomas (67%). STAT3 has not been previously investigated in plasmablastic lymphoma and our findings suggest that this pathway may be also relevant in these tumors. The expression of pSTAT3 observed in our subset of non-germinal center B-cell-like diffuse large B-cell lymphomas is similar to the frequency observed in previous studies.45, 46 Although the prognostic significance of this finding in diffuse large B-cell lymphoma is controversial,47, 48 studies in cell lines and animal models suggest that STAT3 may be a relevant therapeutic target for this subgroup of diffuse large B-cell lymphomas.45, 46, 49
The high expression of pSTAT3 in ALK-positive large B-cell lymphomas may be relevant for the plasmablastic phenotype of these tumors since STAT3 promotes the upregulation of BLIMP1, a master regulator of the plasma cell differentiation program.21 Previous studies have characterized the phenotype of ALK-positive large B-cell lymphomas with CD138 positivity and lack of mature B-cell markers such as CD20 and CD79a, but the expression of BLIMP1 and other markers of the plasma cell differentiation program was not known. Consistent with the role of pSTAT3, we have demonstrated that ALK-positive large B-cell lymphomas constantly express BLIMP1 and it seems to be functional because the tumors lack PAX5 and express XBP1, two genes, respectively, repressed and upregulated by BLIMP1. PAX5 is essential to maintain the mature B-cell identity and its repression is required for the development of the plasma cell differentiation program, whereas XBP1 is an important transcription factor that modulates the terminal B-cell differentiation program.50, 51 This complete plasmablastic phenotype observed in ALK-positive large B-cell lymphomas is less common in plasmablastic lymphomas. These tumors constantly express BLIMP1 in virtually all cases, but XBP1 is only expressed in approximately 50% of them.52, 53
MYC rearrangements have been observed in 50% of plasmablastic lymphomas and plasmablastic plasma cell myelomas. BLIMP1, constantly expressed in these tumors, represses genes related to cell proliferation and growth including MYC.54 The high frequency of MYC rearrangements in these tumors may be an oncogenic mechanism to overcome the repressor effect of BLIMP1 on MYC expression.25 In this study, we have observed that, contrary to plasmablastic lymphomas, ALK-positive large B-cell lymphomas do not have MYC rearrangements, although one case had MYC amplifications. In spite of the low incidence of genetic alterations, MYC protein was expressed in all tumors, suggesting that it may be important for the pathogenesis of the tumor. Previous studies have shown that MYC is downstream of ALK55 and may also be a target of activated STAT3.56, 57 Therefore, MYC expression in ALK-positive large B-cell lymphomas may be driven by these alternative mechanisms that, as postulated for plasmablastic lymphomas, may lead to overcome the repressor effect of BLIMP1.
In conclusion, our study shows that ALK-positive large B-cell lymphomas express markers associated with a complete plasma cell differentiation program and active STAT3, but contrary to other aggressive neoplasms with plasmablastic phenotype lack MYC rearrangements. These findings are consistent with a model in which ALK rearrangements activate STAT3 that in turn promotes the plasma cell differentiation program through BLIMP1, and upregulates the expression of MYC (Figure 3). The relevance of the ALK/STAT3 pathway in the pathogenesis of these lymphomas may offer an attractive target for new therapies.

Model of signal transducers and activators of transcription-3 (STAT3) activation and BLIMP1 and MYC expression in ALK+ large B-cell lymphoma (LBCL). ALK rearranged receptor bound to a non-rearranged ALK receptor (ALK wild type, ALK WT) triggers a STAT3 homodimer formation without the presence of an external ligand. STAT3 dimerization results in the phosphorylation of its tyrosine residues. This activation allows the entrance of the STAT3 dimer into the nucleus of the cell, where it promotes the activation of BLIMP1, which jointly with interferon regulatory factor-4 (IRF4) and X-box binding protein 1 (XBP1) trigger the plasma cell differentiation, and the activation of MYC in the cell proliferation.
References
Delsol G, Lamant L, Mariame B et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2; 5 translocation. Blood 1997;89:1483–1490.
Gascoyne RD, Lamant L, Martin-Subero JI et al. ALK-positive diffuse large B-cell lymphoma is associated with Clathrin-ALK rearrangements: report of 6 cases. Blood 2003;102:2568–2573.
Beltran B, Castillo J, Salas R et al. ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature. J Hematol Oncol 2009;2:11.
Lee HW, Kim K, Kim W et al. ALK-positive diffuse large B-cell lymphoma: report of three cases. Hematol Oncol 2008;26:108–113.
Ott G, Bastian BC, Katzenberger T et al. A lymphohistiocytic variant of anaplastic large cell lymphoma with demonstration of the t(2;5)(p23;q35) chromosome translocation. Br J Haematol 1998;100:187–190.
Adam P, Katzenberger T, Seeberger H et al. A case of a diffuse large B-cell lymphoma of plasmablastic type associated with the t(2;5)(p23;q35) chromosome translocation. Am J Surg Pathol 2003;27:1473–1476.
Cerchietti L, mm-Welk C, Vater I et al. Inhibition of anaplastic lymphoma kinase (ALK) activity provides a therapeutic approach for CLTC-ALK-positive human diffuse large B cell lymphomas. PLoS One 2011;6:e18436.
De Paepe P, Baens M, van KH et al. ALK activation by the CLTC-ALK fusion is a recurrent event in large B-cell lymphoma. Blood 2003;102:2638–2641.
Ishii K, Yamamoto Y, Nomura S . CD30-negative diffuse large B-cell lymphoma expressing ALK. Rinsho Ketsueki 2005;46:501–506.
Onciu M, Behm FG, Downing JR et al. ALK-positive plasmablastic B-cell lymphoma with expression of the NPM-ALK fusion transcript: report of 2 cases. Blood 2003;102:2642–2644.
Shi M, Miron PM, Hutchinson L et al. Anaplastic lymphoma kinase-positive large B-cell lymphoma with complex karyotype and novel ALK gene rearrangements. Hum Pathol 2011;42:1562–1567.
Stachurski D, Miron PM, Al-Homsi S et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma with a complex karyotype and cryptic 3′ ALK gene insertion to chromosome 4 q22-24. Hum Pathol 2007;38:940–945.
Van Roosbroeck K, Cools J, Dierickx D et al. ALK-positive large B-cell lymphomas with cryptic SEC31A-ALK and NPM1-ALK fusions. Haematologica 2010;95:509–513.
Zhang D, Denley RC, Filippa DA et al. ALK-positive diffuse large B-cell lymphoma with the t(2;17)(p23;q23). Appl Immunohistochem Mol Morphol 2009;17:172–177.
Chiarle R, Voena C, Ambrogio C et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008;8:11–23.
Mosse YP, Wood A, Maris JM . Inhibition of ALK signaling for cancer therapy. Clin Cancer Res 2009;15:5609–5614.
Chiarle R, Gong JZ, Guasparri I et al. NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 2003;101:1919–1927.
Kuefer MU, Look AT, Pulford K et al. Retrovirus-mediated gene transfer of NPM-ALK causes lymphoid malignancy in mice. Blood 1997;90:2901–2910.
Chiarle R, Simmons WJ, Cai H et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med 2005;11:623–629.
Zamo A, Chiarle R, Piva R et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene 2002;21:1038–1047.
Diehl SA, Schmidlin H, Nagasawa M et al. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol 2008;180:4805–4815.
Montes-Moreno S, Montalban C, Piris MA . Large B-cell lymphomas with plasmablastic differentiation: a biological and therapeutic challenge. Leuk Lymphoma 2012;53:185–194.
Yu H, Kortylewski M, Pardoll D . Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007;7:41–51.
Taddesse-Heath L, Meloni-Ehrig A, Scheerle J et al. Plasmablastic lymphoma with MYC translocation: evidence for a common pathway in the generation of plasmablastic features. Mod Pathol 2010;23:991–999.
Valera A, Balague O, Colomo L et al. IG/MYC rearrangements are the main cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol 2010;34:1686–1694.
Avet-Loiseau H, Gerson F, Magrangeas F et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001;98:3082–3086.
Green TM, Nielsen O, de SK et al. High levels of nuclear MYC protein predict the presence of MYC rearrangement in diffuse large B-cell lymphoma. Am J Surg Pathol 2012;36:612–619.
Kluk MJ, Chapuy B, Sinha P et al. Immunohistochemical detection of MYC-driven diffuse large B-cell lymphomas. PLoS One 2012;7:e33813.
Ruzinova MB, Caron T, Rodig SJ . Altered subcellular localization of c-Myc protein identifies aggressive B-cell lymphomas harboring a c-MYC translocation. Am J Surg Pathol 2010;34:882–891.
Tapia G, Lopez R, Munoz-Marmol AM et al. Immunohistochemical detection of MYC protein correlates with MYC gene status in aggressive B cell lymphomas. Histopathology 2011;59:672–678.
Swerdlow SH, Campo E, Harris N et al WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, France, 2008.
Gutierrez-Garcia G, Cardesa-Salzmann T, Climent F et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood 2011;117:4836–4843.
Ventura RA, Martin-Subero JI, Jones M et al. FISH analysis for the detection of lymphoma-associated chromosomal abnormalities in routine paraffin-embedded tissue. J Mol Diagn 2006;8:141–151.
Bubala H, Maldyk J, Wlodarska I et al. ALK-positive diffuse large B-cell lymphoma. Pediatr Blood Cancer 2006;46:649–653.
Chikatsu N, Kojima H, Suzukawa K et al. ALK+, CD30−. Mod Pathol 2003;16:828–832.
Gesk S, Gascoyne RD, Schnitzer B et al. ALK-positive diffuse large B-cell lymphoma with ALK-Clathrin fusion belongs to the spectrum of pediatric lymphomas. Leukemia 2005;19:1839–1840.
Isimbaldi G, Bandiera L, d'Amore ES et al. ALK-positive plasmablastic B-cell lymphoma with the clathrin-ALK gene rearrangement. Pediatr Blood Cancer 2006;46:390–391.
Laurent C, Do C, Gascoyne RD et al. Anaplastic lymphoma kinase-positive diffuse large B-cell lymphoma: a rare clinicopathologic entity with poor prognosis. J Clin Oncol 2009;27:4211–4216.
Li S . Anaplastic lymphoma kinase-positive large B-cell lymphoma: a distinct clinicopathological entity. Int J Clin Exp Pathol 2009;2:508–518.
McManus DT, Catherwood MA, Carey PD et al. ALK-positive diffuse large B-cell lymphoma of the stomach associated with a clathrin-ALK rearrangement. Hum Pathol 2004;35:1285–1288.
Reichard KK, McKenna RW, Kroft SH . ALK-positive diffuse large B-cell lymphoma: report of four cases and review of the literature. Mod Pathol 2007;20:310–319.
Takeuchi K, Soda M, Togashi Y et al. Identification of a novel fusion, SQSTM1-ALK, in ALK-positive large B-cell lymphoma. Haematologica 2011;96:464–467.
Barreca A, Lasorsa E, Riera L et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol 2011;47:R11–R23.
Momose S, Tamaru J, Kishi H et al. Hyperactivated STAT3 in ALK-positive diffuse large B-cell lymphoma with clathrin-ALK fusion. Hum Pathol 2009;40:75–82.
Ding BB, Yu JJ, Yu RY et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008;111:1515–1523.
Lam LT, Wright G, Davis RE et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008;111:3701–3713.
Gupta M, Maurer MJ, Wellik LE et al. Expression of Myc but not pSTAT3, is an adverse prognostic factor for diffuse large cell lymphoma treated with epratuzumab/R-CHOP. Blood 2012;120:4400–4406.
Wu ZL, Song YQ, Shi YF et al. High nuclear expression of STAT3 is associated with unfavorable prognosis in diffuse large B-cell lymphoma. J Hematol Oncol 2011;4:31.
Scuto A, Kujawski M, Kowolik C et al. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res 2011;71:3182–3188.
Klein U, la-Favera R . Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol 2008;8:22–33.
Reimold AM, Iwakoshi NN, Manis J et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001;412:300–307.
Balague O, Mozos A, Martinez D et al. Activation of the endoplasmic reticulum stress-associated transcription factor × box-binding protein-1 occurs in a subset of normal germinal-center B cells and in aggressive B-cell lymphomas with prognostic implications. Am J Pathol 2009;174:2337–2346.
Montes-Moreno S, Gonzalez-Medina AR, Rodriguez-Pinilla SM et al. Aggressive large B-cell lymphoma with plasma cell differentiation: immunohistochemical characterization of plasmablastic lymphoma and diffuse large B-cell lymphoma with partial plasmablastic phenotype. Haematologica 2010;95:1342–1349.
Lin Y, Wong K, Calame K . Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 1997;276:596–599.
Raetz EA, Perkins SL, Carlson MA et al. The nucleophosmin–anaplastic lymphoma kinase fusion protein induces c-Myc expression in pediatric anaplastic large cell lymphomas. Am J Pathol 2002;161:875–883.
Han SS, Yun H, Son DJ et al. NF-kappaB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol Cancer 2010;9:97.
Sarosiek KA, Malumbres R, Nechushtan H et al. Novel IL-21 signaling pathway up-regulates c-Myc and induces apoptosis of diffuse large B-cell lymphomas. Blood 2010;115:570–580.
Acknowledgements
We thank Elena Gonzalvo, Laura Gelabert and Mònica Marín for their excellent technical assistance. This study was funded by ‘Comisión Interministerial de Ciencia y Tecnología Española’ (CICYT) SAF 12/38432, Red Temática de Investigación Cooperativa del Cáncer (RTICC) (RD12/0036/0086) and ‘Conveni Programa de Feno/Genotipatge per al diagnòstic i tractament individualitzat del pacient oncològic, La Caixa’. Figure 3 was produced using Servier Medical Art (http: //www.servier.com/).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Valera, A., Colomo, L., Martínez, A. et al. ALK-positive large B-cell lymphomas express a terminal B-cell differentiation program and activated STAT3 but lack MYC rearrangements. Mod Pathol 26, 1329–1337 (2013). https://doi.org/10.1038/modpathol.2013.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2013.73
Keywords
This article is cited by
-
Plasmablastic lymphoma occurring in ulcerative colitis during treatment with immunosuppressive therapy
Clinical Journal of Gastroenterology (2023)
-
Diffuse large B-cell lymphomas, not otherwise specified, and emerging entities
Virchows Archiv (2023)
-
Plasmablastic lymphoma phenotype is determined by genetic alterations in MYC and PRDM1
Modern Pathology (2017)
-
The clinicopathologic spectrum of mature aggressive B cell lymphomas
Virchows Archiv (2017)
