
Abstract
Mast cell sarcoma is a rare, aggressive neoplasm composed of cytologically malignant mast cells presenting as a solitary mass. Previous descriptions of mast cell sarcoma have been limited to single case reports, and the pathologic features of this entity are not well known. Here, we report three new cases of mast cell sarcoma and review previously reported cases. Mast cell sarcoma has a characteristic morphology of medium-sized to large epithelioid cells, including bizarre multinucleated cells, and does not closely resemble either normal mast cells or the spindle cells of systemic mastocytosis. One of our three cases arose in a patient with a remote history of infantile cutaneous mastocytosis, an association also noted in one previous case report. None of our three cases were correctly diagnosed as mast cell neoplasms on initial pathological evaluation, suggesting that this entity may be under-recognized. Molecular testing of mast cell sarcoma has not thus far detected the imatinib-resistant KIT D816V mutation, suggesting that recognition of these cases may facilitate specific targeted therapy.
Similar content being viewed by others

Main
Mast cell neoplasms comprise a clinically and biologically heterogeneous group of disorders.1 The most common of these are clinically indolent clonal mast cell proliferations predominantly involving the skin (cutaneous mastocytosis) and bone marrow (systemic mastocytosis), whose primary symptomatic manifestations are due to the unique paracrine and systemic effects of mast cell secretory products. Aggressive mast cell neoplasms are rare and are typically variants of systemic mastocytosis that diffusely involve the bone marrow and other anatomic sites, in some cases associated with progression to mast cell leukemia, or the development of a non-mast cell hematologic neoplasm. Although relatively common in dogs,2 mast cell sarcoma, defined as a malignant mast cell neoplasm presenting as an isolated destructive mass, is exceedingly rare in humans.1 As only seven cases of mast cell sarcoma have been reported to date in the English language literature,3, 4, 5, 6, 7, 8, 9 each representing a single case report, there is limited information available regarding the diagnostic features, clinical behavior, and genetic aberrations associated with this entity.
Here, we present three new cases of mast cell sarcoma, and place them in context with the previously reported cases. We find that mast cell sarcoma may present in a broad spectrum of anatomic locations and age groups. The cells of mast cell sarcoma are medium to large, often bizarre-appearing epithelioid cells with characteristic morphologic features and a specific immunophenotype. However, because they bear only limited resemblance to normal mast cells, none of our cases were correctly diagnosed on initial pathological evaluation. We note that KIT genotyped cases of mast cell sarcoma in our series and prior case reports have not demonstrated the imatinib-resistant KIT D816V mutation, but rather showed either an absence of KIT mutations, or mutations in KIT domains associated with imatinib sensitivity in other neoplasms. These findings suggest that accurate diagnosis of mast cell sarcoma may allow for specific targeted therapy for this aggressive malignancy.
Materials and methods
Cases of mast cell sarcoma were retrieved from the consultation files of two of the authors (J.A.F. and J.L.H). Formalin-fixed, paraffin-embedded tissue was stained immunohistochemically using the antibodies listed in Table 1.
Tissue for electron microscopy was extracted from formalin-fixed, paraffin embedded tissue blocks, soaked in 100% xylene overnight, rehydrated in a series of ethanol solutions, rinsed in sodium cacodylate buffer, and fixed for 1.5 h with 2.5% glutaraldehyde, 2.0% paraformaldehyde, and 0.025% calcium chloride, in a 0.1 M sodium cacodylate buffer, pH 7.4. Tissues were further processed in a Leica Lynx automatic tissue processor. Briefly, tissues were post fixed with osmium tetroxide, dehydrated in a series of ethanol solutions, en bloc stained during the 70% ethanol dehydration step for one hour, infiltrated with propylene oxide epoxy mixtures, embedded in pure epoxy, and polymerized over night at 60 °C. Thin sections were stained with lead citrate and examined with an FEI Morgagni transmission electron microscope. Images were captured with an AMT (Advanced Microscopy Techniques) digital CCD camera.
The study protocols were approved by the Institutional Review Board of Partners Healthcare.
Results
Patient 1
Patient 1 was a 12-year-old female who initially presented with a large left middle ear mass. A biopsy of the mass was felt to be consistent with Langerhans cell histiocytosis. The tumor progressed intracranially, despite systemic treatment with vinblastine and prednisone. Two debulking surgeries were performed. Unfortunately, subsequent radiation therapy did not produce a response, and the patient developed local progression involving the skull.
At this time, the pathology was reviewed at a second institution, and an unspecified histiocytic or myeloid neoplasm was favored. She was treated with multiple courses of chemotherapy, including 2CDA/Ara-C, ICE, clofarabine, ALCL 99, idarubicin/ velcade/Ara-C, and decitabine over 27 months following initial presentation. Although the tumor showed an initial response, there was subsequent intracranial progression near the left sphenoid and transverse venous sinus.
The pathology was then reviewed at a third institution, and a conclusive diagnosis of a malignant mast cell neoplasm was rendered. Of note, a total of five bone marrow biopsies had been performed in the 20 months following initial diagnosis, all of which were negative for involvement by systemic mastocytosis, even on retrospective review. The patient was treated with radiation and imatinib mesylate, and scheduled for allogeneic hematopoietic stem cell transplantation. Serum tryptase levels have remained markedly elevated (ranging from 122 ng/ml to >200). At last follow-up, the patient is alive with persistent disease 45 months following initial presentation.
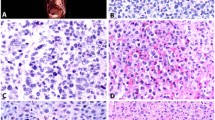
Histologically, all biopsies and excision specimens showed similar morphologic findings (Figure 1). There was a dense proliferation of morphologically heterogeneous, medium-sized to large cells, with well-defined cell borders, clear to palely eosinophilic cytoplasm, and irregular nuclei. A prominent, patchy infiltrate of eosinophils was also present. Scattered very large epithelioid cells were also present; these often showed bizarre features, including multilobated nuclei and multinucleation. There was occasional emperipolesis of eosinophils within the large cells. Mitotic activity varied from 2 per 10 high power fields in the initial specimen to 18 per 10 high power fields in a recurrence.

Representative histological images from the intracranial tumor in case 1. Note the dense infiltration of eosinophils, tumor cells with well-defined cell borders and lobulated nuclei, and scattered multinucleated tumor cells (a–b). The tumor cells show variable expression of KIT (c), and strong membranous CD25 (d). Mast cell tryptase was also positive (not shown).
Sequencing of the KIT gene in tumor tissue showed no evidence of a mutation. Immunohistochemical findings for all three cases are summarized in Table 2.
Patient 2
Patient 2 was a 19 year-old male who presented with a progressively enlarging submucosal nodule on the right inner aspect of his lower lip. His history was notable for childhood-onset cutaneous mastocytosis, which had lasted from 1 month to approximately 2 years of age, including blistering lesions on the chest and upper trunk. The lesions were reportedly treated with clobetasol ointment, and healed without scarring or residual hyperpigmentation. The patient reported having a tan-colored lesion on his right lip for his entire childhood, which was stable until a few months before presentation. The patient denied urticaria or flushing, but did report recurrent periods of nausea, vomiting and diarrhea as well as a 50-pound weight loss. An excisional biopsy of the lesion was performed.
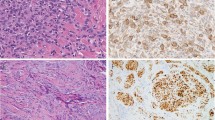
Histologically, the excision reveled a discrete, densely cellular mass extending from the deep dermis into subcutaneous tissues, with destructive invasion of minor salivary glands and skeletal muscle. The tumor was composed of large, pleomorphic epithelioid cells with abundant finely granular eosinophilic cytoplasm (Figure 2a). Some cells showed a more uniformly eosinophilic cytoplasm, while other cells also contained large clear vacuoles especially at the periphery, and some contained densely eosinophilic perinuclear aggregates imparting a vaguely rhabdoid appearance. Scattered very large cells with bizarre nuclear features were present, including forms with bilobed, horseshoe-shape, and multilobulated nuclei. The nuclei of most cells showed round to moderately irregular contours and coarse chromatin with numerous basophilic chromocenters, and usually solitary eosinophilic nucleoli. Mitoses, including atypical forms, were readily identified, up to 16 per 10 high-power fields. Scattered karyorrhectic tumor cells were also present. Morphologically normal eosinophils were scattered throughout the tumor. Large cells were absent from the superficial dermis, but this area was involved by an infiltrate of numerous morphologically typical interstitial mast cells, consistent with the findings of urticaria pigmentosa. The excisional margin was involved by tumor cells.

Representative histological images from the lip tumor in case 2. Tumor cells show well-defined borders, marked anisocytosis, and round to multilobulated nuclei (a). Giemsa stain reveals scant, fine metachromatic granules in many cells, as well as scattered prominently granulated cells (b). The sarcoma was strongly KIT positive, and largely involved the deep dermis and subcutaneous tissue. However, there was also a diffusely increased population of morphologically typical, KIT-positive mast cells within the superficial dermis, consistent with residual urticaria pigmentosa (UP) (c). The highly pleomorphic mast cells of the sarcomatous component contrast with the benign-appearing mast cells of the UP component (arrows) in this field from the tumor edge (d).
The initial immunohistochemical workup did not include stains for mast cell-specific antigens. A diagnosis of ALK-negative anaplastic large cell lymphoma was favored by the submitting pathologist. On referral, additional immunostains (Figure 3, Table 2) confirmed a diagnosis of mast cell sarcoma. A Giemsa stain (Figure 2b) revealed the presence of distinctive metachromatic purple granules which were highly variable in density from cell to cell.

Immunohistochemistry in case 2 showed strong tumor cell expression of CD2 (a), CD30 (b), and mast cell tryptase (c). The Ki-67 staining index was approximately 50% (d).
Electron microscopy performed on formalin-fixed tissue retrieved from paraffin demonstrated numerous vacuoles in most cells. Many of the vacuoles were empty (degranulated), but scattered cells had characteristic mast cell features (Figure 4),10 containing intracytoplasmic membrane-bound granules with electron-dense material or finely granular material, as well as some granules with a suggestion of lamellar structures. KIT gene mutation screening revealed the presence of a deletion mutation in exon 8 (D419del).

Electron microscopic examination of case 2 demonstrated that the neoplastic cells contained numerous intracytoplasmic membrane-bound granules with electron-dense material (single arrow), particulate content (arrowhead), and a suggestion of lamellar structures (double arrows).
Subsequently a serum tryptase level was found to be elevated at 18 ng/ml. A bone marrow biopsy showed a mildly hypocellular marrow with maturing trilineage hematopoiesis and 2% mast cells, including occasional spindled forms and small clusters of mast cells (<15 cells per aggregate). A small subset of the mast cells was positive for CD25 by immunohistochemistry and flow cytometry. Overall, although the W.H.O. criteria were not fulfilled, the findings were suspicious for limited involvement by systemic mastocytosis.
The patient was started on imatinib, 400 mg daily. Re-excision of the surgical site seven months after the initial excision showed morphologically normal, but quantitatively increased mast cells, consistent with residual urticaria pigmentosa; there was no evidence of residual sarcoma. CT scans of the head, neck, chest, abdomen, and pelvis showed no suspicious lesions at eight months post excision, and serum tryptase levels were within the normal range at nine and twelve months post-excision. At 19 months post-excision, an enlarged cervical lymph node was detected, and biopsy demonstrated recurrent mast cell sarcoma.
Patient 3
Patient 3 was a 77 year-old woman who presented with a six-month history of progressively worsening pain in her right hip and groin. She had a history of nephrolithiasis and cystectomy for chronic cystitis, as well as chronic anemia. Her family history was significant for ovarian cancer, an unspecified brain tumor, and melanoma. A pelvic CT scan initially showed no identifiable lesion at the site of the pain (Figure 5a). Six months later, an MRI scan demonstrated a pelvic mass, which was now clearly visible by CT scan as a destructive lesion arising within the right supra-acetabular pelvis, and extending into the adjacent soft tissues (Figure 5b). A bone scan showed markedly increased signal at the site of the right pelvic mass, but no evidence of disease at other sites (Figure 5c). A core needle biopsy was performed, but no definitive diagnosis was made on initial pathology evaluation, and she was treated with 30 Gy of external beam radiation to the mass as a palliative measure. Subsequently, pathological consultation and additional immunohistochemistry yielded a conclusive diagnosis of mast cell sarcoma.

Pelvic CT scans of patient 3 at the time of diagnostic biopsy (b), and 6 months prior (a) demonstrate rapid growth of a destructive mass arising above the right acetabulum. A bone scan demonstrated abnormal signal localized to the right pelvis (c). The tumor cells showed histological features similar to those of the other two cases (d), and were variably positive for mast cell tryptase (e).
Histologically, the tumor was composed of sheets of highly pleomorphic epithelioid cells, ranging from medium-sized cells with round nuclei to large cells with multiple or lobulated nuclei (Figure 5d). The cytoplasm was eosinophilic, granular, and variably vacuolated. Morphologically unremarkable eosinophils were scattered throughout the tumor. A Giemsa stain showed a fine scattering of metachromatic purple granules in some tumor cells, while others showed similar granules in dense perinuclear clumps. Stains for KIT and mast cell tryptase were strongly positive.
Following establishment of the diagnosis, the patient was found to have an elevated serum tryptase at 28.2 ng/ml. The patient declined a bone marrow biopsy. She was briefly treated with pamidronate. A CT scan showed a slight interval decrease in the right pelvic mass, from 8.5 × 6 cm to 7.6 × 5 cm, two months after completion of radiotherapy. She subsequently developed a urinary tract infection resistant to multiple antibiotics, declined further therapy, and died under hospice care four months after diagnosis.
Discussion
Mast cell sarcoma is defined in the 2008 WHO classification as a variant of mastocytosis that presents as a unifocal mast cell tumor with destructive growth and high-grade cytology. These cases do not fulfill the major criterion for systemic mastocytosis, which requires multifocal, dense infiltrates of mast cells in the bone marrow or other extracutaneous organs. By these criteria, the entire published literature on this entity to date consists of seven case reports, the clinical and histological characteristics of which are compared to the current three cases in Table 3.
The age at presentation for these cases ranges from 4 to 77 years, similar to the broad age distribution of systemic mastocytosis, but not cutaneous mastocytosis, which typically presents in infancy. The sites of presentation for mast cell sarcoma are also diverse, with two cases presenting in squamous mucosal sites (lip and subglottis), two in the gastrointestinal tract, two involving the cranial bones and meninges, two in the extracranial skeleton, one in the uterus, and one in the skin. This distribution may be related to the normal tissue distribution of mast cells in mucosal sites and bone marrow.
Despite their diverse sites of presentation, the three cases described in our study showed similar histological features, which were distinct from typical cases of systemic mastocytosis, but were shared by many of the previously reported cases of mast cell sarcoma. The majority of tumor cells in our cases were medium-sized to large epithelioid cells with abundant cytoplasm and well-defined cell borders. Cells with prominent nuclear irregularities and bilobation, as well as bizarre multinucleated tumor giant cells were prominent in our cases, and were reported in most of the published cases as well. Thus, the cells of mast cell sarcoma do not closely resemble either normal tissue mast cells or the spindled mast cells typical of most systemic mastocytosis cases, but are similar to the cytologically defined ‘atypical type II mast cells’ or ‘promastocytes’ reported in some cases of aggressive systemic mastocytosis.11 Neoplastic mast cells with lobulated nuclei have been described in histologic sections from cases of aggressive systemic mastocytosis,1 including at least one case which progressed to include sarcoma-like mass lesions.12 ‘Metachromatic blasts’ with a high nuclear-to-cytoplasmic ratio have been reported in association with mast cell leukemia, but were not a feature of our three cases, although several of the previously reported cases of mast cell sarcoma did progress to mast cell leukemia and showed a more blast-like morphology at that time. An important morphologic clue linking these cases to systemic mastocytosis was the presence of infiltrating eosinophils, although this finding is certainly shared with other entities, as highlighted by the initial misdiagnosis of case 1 as Langerhans cell histiocytosis.
It is not surprising that none of the three cases of mast cell sarcoma were correctly diagnosed as mast cell neoplasms at the time of initial pathological evaluation, given their lack of morphological resemblance to normal mast cells, or to more familiar mast cell neoplasms. All three cases were initially evaluated with broad immunohistochemical panels that did not include specific mast cell markers. Case 2 highlights the potential immunophenotypic pitfalls of this entity, as this tumor was positive for antigens commonly associated with histiocytic or myeloid sarcoma (CD68, CD117, weak lysozyme), anaplastic large cell lymphoma (CD2, CD4, CD25, CD30, CD43), and melanoma (MITF). MITF is a transcription factor that is known to be expressed in normal mast cells,13 but is not often used as a marker for this lineage in diagnostic practice.
Diagnostic considerations suggested by referring pathologists in our cases included poorly differentiated carcinoma, melanoma, ALK-negative anaplastic large cell lymphoma, Langerhans cell histiocytosis, and other myeloid or histiocytic neoplasms. Like our mast cell sarcoma cases, Langerhans cell histiocytosis typically shows tumor cells with abundant pink cytoplasm, and a dense infiltrate of eosinophils. However, the characteristic longitudinal nuclear grooves of Langerhans cell histiocytosis were not seen in our mast cell sarcoma cases, and essentially all cases of Langerhans cell histiocytosis should show immunohistochemical expression of S100, CD1a, and langerin. The well-defined cytoplasmic borders seen in mast cell sarcoma also contrasts with the indistinct borders seen in most cases of Langerhans cell histiocytosis and histiocytic sarcoma. Like our mast cell sarcoma cases, anaplastic large cell lymphoma typically demonstrates sheet-like growth of large epithelioid cells which often show bilobed or multilobated nuclei. Abundant eosinophils have been reported in anaplastic large cell lymphoma, but are relatively uncommon,14 and should prompt consideration of other diagnoses. Variable KIT expression may occasionally be seen in T cell lymphomas, but the uniformly strong expression seen in two of our three mast cell sarcoma cases would be highly unusual, and expression of CD68 should not be seen. Because no single immunohistochemical marker is specific for ALK-negative anaplastic large cell lymphoma, it may be prudent to exclude mast cell sarcoma before making this diagnosis in cases lacking expression of CD3, CD5, EMA, or cytotoxic granule markers, or when PCR studies fail to demonstrate a clonal T cell receptor rearrangement.
Both our cases and those from the literature support mast cell tryptase as a highly specific stain for confirming the lineage of mast cell sarcoma, once the diagnosis has been considered. Giemsa stain was also positive, albeit in a subset of cells; metachromatic granules were not universally detected in mast cell sarcoma among the previously reported cases. While it has recently been reported that CD30 immunohistochemistry may be helpful in distinguishing between indolent and aggressive cases of systemic mastocytosis,15 this marker may be less helpful in mast cell sarcoma, as it was only positive in 1 of 2 evaluated cases from our series, and was negative in all three of the prior mast cell sarcoma cases for which it was reported.
KIT sequencing results were only available for two of our cases, due to tissue exhaustion of the very small biopsy from case 3, and the patient’s decision to decline a repeat biopsy. However, among the five cases of mast cell sarcoma screened for the presence of a KIT mutation in our series and the literature, it is striking to note that none demonstrated the canonical D816V mutation of the KIT kinase domain, despite the fact that this mutation has been reported in >95% of cases of adult systemic mastocytosis.1 Two mast cell sarcoma cases showed alternate KIT mutations, while no KIT mutation was detected in the sequenced exons from the other 3 cases. While the sensitivity of direct sequencing of bone marrow aspirates from typical cases of systemic mastocytosis may be compromised due to a dilution by normal bone marrow cellular elements, depending on the molecular method employed, this seems unlikely to explain the lack of detected mutations in these three cases, given the high tumor cell content of mast cell sarcoma. This observation is not merely of academic interest, as mast cell neoplasms with the D816V mutation are resistant to therapy with imatinib and some other clinically available KIT tyrosine kinase inhibitors.16 In contrast, cases of mastocytosis that lacked KIT D816V mutations have been effectively treated with tyrosine kinase inhibitors, including a case of pediatric cutaneous mastocytosis that responded to imatinib and had a mutation identical to that found in our mast cell sarcoma case 2.17 In the case of uterine mast cell sarcoma reported by Ma et al., which lacked a detectable KIT mutation, the patient achieved a durable clinical response with imatinib therapy, and in our case 2, which contained a known imatinib-sensitive KIT mutation, initiation of imatinib therapy coincided with a decrease of the patient’s serum tryptase to the normal range and a tumor-free interval of well over a year, although recurrent disease was subsequently detected in a regional lymph node.
There is limited evidence for efficacy of other therapies in mast cell sarcoma. Radiation therapy failed to produce a response in our Case 1, and produced a minimal radiologic response in our Case 3; this is consistent with the limited efficacy noted in other case reports. Radiation therapy has been shown to be ineffective at reducing mast cell numbers or activation in vitro,18 suggesting that this treatment modality may not be adequate to control the expansion of mast cell sarcomas. In our patient 1, the use of cytotoxic chemotherapy regimens appropriate for myeloid or lymphoid neoplasia produced only transient responses, consistent with other literature reports.
Our case 2 is of particular interest in that the tumor occurred in a patient who had recovered from infantile cutaneous mastocytosis 17 years previously, and apparently arose in a residual lesion of urticaria pigmentosa in the mucosa of the lip, a clinical evolution similar to that of the case recently reported by Auquit-Auckbur et al.9 Interestingly, case 2 showed urticaria pigmentosa and mast cell sarcoma in two different regions of the same biopsy. These two cases appear to represent malignant transformation of a previously quiescent mast cell clone, implying that at least some cases of mast cell sarcoma may be biologically more closely related to pediatric cutaneous mastocytosis than to adult systemic mastocytosis. This hypothesis is supported by the fact that the exon 8 KIT mutation seen in case 2 is more characteristic of pediatric cutaneous mastocytosis than adult systemic mastocytosis.19 There are at least two prior reports of cutaneous mastocytomas with atypical cytological features,20, 21 including occasional neoplastic mast cells with bilobed nuclei, but the cases of cutaneous mast cell sarcoma differed from these in showing frankly malignant cytomorphology, with a high degree of tumor cell anaplasia, numerous mitoses (including atypical forms), and a relatively high Ki-67 index.
In summary, mast cell sarcoma is an aggressive mast cell neoplasm with clinical, histological, and genetic features distinct from other forms of mastocytosis, and which may rarely arise due to late malignant transformation of cutaneous mastocytosis. Although this diagnosis is readily confirmed with specific immunohistochemical markers, its rarity and anaplastic morphology present a diagnostic challenge. Given the specific therapeutic possibilities available to patients with this entity, including KIT tyrosine kinase inhibitors and therapies optimized for other aggressive myeloid neoplasms, it is essential for pathologists to accurately diagnose this rare tumor type.
Author's note added in proof
Following acceptance of this manuscript, two additional cases of mast cell sarcoma were described in a report by Georgin-Lavialle et al. (Georgin-Lavialle et al. Mast cell sarcoma: a rare and aggressive entity-report of two cases and review of the literature. J Clin Oncol 2012 Nov 5. (E-pub ahead of print) PMID: 23129735). One tumor arose in a patient with a longstanding history of familial indolent mastocytosis with urticaria pigmentosa. The second case was initially misdiagnosed as anaplastic large cell lymphoma (due to CD30 expression), and later showed a transient partial response to steroids and dasatinib. Both cases lacked the KIT D816V mutation; the latter case harbored a KIT exon 11 mutation (p. Val560Gly). Both tumors showed an aggressive clinical course; the patients died 6 months and 13 months after initial diagnosis.
References
Horny H-P, Metcalfe DD, Bennett JM et al. In: Swerdlow SH, Campo E, Harris NL, et al (eds) WHO classification of tumors of haematopoietic and lymphoid tissues 4th edn. International Agency for Research on Cancer: Lyon, 2008, pp 54–63.
Patnaik AK, Ehler WJ, MacEwan EG . Canine cutaneous mast cell tumor: morphologic grading and survival time in 83 dogs. Vet Pathol 1984;21:469–474.
Horny HP, Parwaresch MR, Kaiserling E et al. Mast cell sarcoma of the larynx. J Clin Pathol 1986;39:596–602.
Kojima M, Nakamura S, Itoh H et al. Mast cell sarcoma with tissue eosinophilia arising in the ascending colon. Mod Pathol 1999;12:739–743.
Chott A, Guenther P, Huebner A et al. Morphologic and immunophenotypic properties of neoplastic cells in a case of mast cell sarcoma. Am J Surg 2003;27:1013–1019.
Brcić L, Vuletić LB, Stepan J et al. Mast-cell sarcoma of the tibia. J Clin Pathol 2007;60:424–425.
Bugalia A, Abraham A, Balasubramanian P . Mast cell sarcoma of the small intestine: a case report. J Clin Pathol 2011;64:1035–1037.
Ma HB, Xu X, Liu WP et al. Successful treatment of mast cell sarcoma of the uterus with imatinib. Int J Hematol 2011;94:491–494.
Auquit-Auckbur I, Lazar C, Deneuve S et al. Malignant transformation of mastocytoma developed on skin mastocytosis into cutaneous mast cell sarcoma. Am J Surg Pathol 2012;36:779–782.
Kobayasi T, Midtgård K, Asboe-Hansen G . Ultrastructure of human mast-cell granules. J Ultrastruct Res 1968;23:153–165.
Sperr WR, Escribano L, Jordan J et al. Morphologic properties of neoplastic mast cells: delineation of stages of maturation and implication for cytological grading of mastocytosis. Leuk Res 2001;25:529–536.
Fine HF, Akin C, Hematti P et al. Presumed choroidal and orbital mastocytosis. Arch Ophthalmol 2001;119:1716–1719.
Takemoto CM, Yoon YJ, Fisher DE . The identification and functional characterization of a novel mast cell isoform of the microphthalmia-associated transcription factor. J Biol Chem 2002;277:30244–30252.
Tilly H, Gaulard P, Lepage E et al. Primary anaplastic large-cell lymphoma in adults: clinical presentation, immunophenotype, and outcome. Blood 1997;90:3727–3734.
Sotlar K, Cerny-Reiterer S, Petat-Dutter K et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol 2011;24:585–595.
Pardanani A, Tefferi A . Systemic mastocytosis in adults: a review on prognosis and treatment based on 342 Mayo Clinic patients and current literature. Curr Opin Hematol 2010;17:125–132.
Hoffmann KM, Moser A, Lohse P et al. Successful treatment of progressive cutaneous mastocytosis with imatinib in a 2-year-old boy carrying a somatic KIT mutation. Blood 2008;112:1655–1657.
Soule BP, Brown JM, Kushnir-Sukhov NM et al. Effects of gamma radiation on FcɛRI and TLR-mediated mast cell activation. J Immunol 2007;179:3276–3286.
Bodemer C, Hermine O, Palmérini F et al. Pediatric mastocytosis is a clonal disease associated with D816V and other activating c-KIT mutations. J Invest Dermatol 2009;130:804–815.
Hu S, Kuo T, Hong HS . Mast cells with bilobed or multilobed nuclei in a nodular lesion of a patient with urticaria pigmentosa. Am J Dermatopathol 2002;24:490.
Tran DT, Jokinen CH, Argenyi ZB . Histiocyte-rich pleomorphic mastocytoma: an uncommon variant mimicking juvenile xanthogranuloma and Langerhans cell histiocytosis. J Cutan Pathol 2009;36:1215–1220.
Guenther PP, Huebner A, Sobottka SB et al. Temporary response of localized intracranial mast cell sarcoma to combination chemotherapy. J Pediatr Hematol Oncol 2001;23:134–138.
Acknowledgements
The authors wish to thank Drs Jenny Kim, Anna Powlowska, William Walsh, George Woodbury, and Steve Covington for providing clinical information regarding patients under their care.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ryan, R., Akin, C., Castells, M. et al. Mast cell sarcoma: a rare and potentially under-recognized diagnostic entity with specific therapeutic implications. Mod Pathol 26, 533–543 (2013). https://doi.org/10.1038/modpathol.2012.199
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.199
Keywords
This article is cited by
-
Mast cell sarcoma of the sternum, clonally related to an antecedent germ cell tumor with a novel D579del KIT mutation
Virchows Archiv (2017)
-
KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis
Leukemia (2015)
-
Primary Mast Cell Disorders in Children
Current Allergy and Asthma Reports (2013)
