
Abstract
Rhabdomyosarcoma is the most common pediatric soft tissue malignancy. Two major subtypes, alveolar rhabdomyosarcoma and embryonal rhabdomyosarcoma, constitute 20 and 60% of all cases, respectively. Approximately 80% of alveolar rhabdomyosarcoma carry two signature chromosomal translocations, t(2;13)(q35;q14) resulting in PAX3–FOXO1 fusion, and t(1;13)(p36;q14) resulting in PAX7–FOXO1 fusion. Whether the remaining cases are truly negative for gene fusion has been questioned. We are reporting the case of a 9-month-old girl with a metastatic neck mass diagnosed histologically as solid variant alveolar rhabdomyosarcoma. Chromosome analysis showed a t(8;13;9)(p11.2;q14;9q32) three-way translocation as the sole clonal aberration. Fluorescent in situ hybridization (FISH) demonstrated a rearrangement at the FOXO1 locus and an amplification of its centromeric region. Single-nucleotide polymorphism-based microarray analysis illustrated a co-amplification of the FOXO1 gene at 13q14 and the FGFR1 gene at 8p12p11.2, suggesting formation and amplification of a chimerical FOXO1–FGFR1 gene. This is the first report to identify a novel fusion partner FGFR1 for the known anchor gene FOXO1 in alveolar rhabdomyosarcoma.
Similar content being viewed by others

Main
Rhabdomyosarcoma accounts for 4–10% of pediatric malignancies. Four pathological subtypes have been traditionally defined: alveolar rhabdomyosarcoma, embryonal rhabdomyosarcoma, botyroid rhabdomyosarcoma, and pleiomorphic rhabdomyosarcoma.1, 2 Alveolar rhabdomyosarcoma and embryonal rhabdomyosarcoma have distinct clinical manifestations, biological behavior, genetic alterations, and account for ∼20 and ∼60% of all cases, respectively.1 Alveolar rhabdomyosarcoma is most often seen in older children and is associated with a poor outcome, while embryonal rhabdomyosarcoma primarily occurs in children younger than 4 years of age and is usually associated with a better prognosis.2 A rare solid variant of alveolar rhabdomyosarcoma has been described that is often morphologically indistinguishable from poorly differentiated embryonal rhabdomyosarcoma and may be confused with other ‘small, round, blue cell tumors’.3
A study of a large pediatric cohort of 171 patients with rhabdomyosarcoma by the children's oncology group (COG) has demonstrated the role of genetics in the development of rhabdomyosarcoma.4 In the subtype of alveolar rhabdomyosarcoma, approximately 80% of cases harbored two signature chromosomal translocations, t(2;13)(q35;q14) in 60%, and t(1;13) (p36;q14) in 20% of cases. These translocations resulted in the formation and overexpression of chimerical genes PAX3–FOXO1 (FOXO1, also known as FKHR) and PAX7–FOXO1, respectively.4 Chromosome rearrangements not involving the FOXO1 locus have also been described in both alveolar rhabdomyosarcoma and embryonal rhabdomyosarcoma.5 In embryonal rhabdomyosarcoma, gains of all or portions of chromosomes 2, 7, 8, 11, 12, 13, 17, 19, and 20, with a particularly high-level gain of chromosome 8 material are consistently observed,6, 7, 8 and loss of heterozygosity (LOH) on the short arm of chromosome 11 (11p15.5) is frequently detected, suggesting that imprinting might be involved.9 In both alveolar rhabdomyosarcoma and embryonal rhabdomyosarcoma, amplification of genes such as MYCN, MDM2, CDK4 and IFG-R1 are often observed.10, 11, 12, 13, 14 Despite the usually sporadic occurrence of rhabdomyosarcoma, it has also been reported in various congenital disorders, including cancer predisposition syndromes and genetic disorders with multi-organ system defects such as Li-Fraumeni syndrome, Beckwith–Wiedemann syndrome, neurofibromatosis type 1, Costello syndrome, Nijmegen breakage syndrome, Rubinstein–Taybi syndrome, Dubowitz syndrome, multiple endocrine neoplasia type 2A, Roberts syndrome, and Duchenne muscular dystrophy.15, 16, 17 Subtype classification and gene fusion status closely correlate with the prognosis.18 However, a question remains, whether alveolar rhabdomyosarcoma cases without PAX3/7–FOXO1 fusion are truly negative for any gene fusion.18, 19, 20, 21 In this study, we describe the clinicopathological assessment, cytogenetic test results, and single nucleotide polymorphism (SNP)-based whole-genome microarray evaluation of a solid variant embryonal rhabdomyosarcoma with a novel FGFR1–FOXO1 fusion and amplification.
Materials and methods
Case Report
A 9-month-old female was admitted with a chief complaint of left neck mass present for a month, and a left shoulder mass present for a week. Chest CT showed a 3 × 1.9 × 2-cm soft tissue mass in the region of the supraspinal muscle, and multiple enlarged cervical lymph nodes. The remainder of the physical examination was within the normal range. The patient underwent excisional biopsy of one of the cervical lymph nodes.
Pathology
Formalin-fixed, paraffin-embedded tissue sections were stained with hematoxylin–phloxine–saffron for routine histological assessment. Immunohistochemical studies were conducted using an automated immunostainer (Thermo Scientific, Freemont, CA, USA). The following primary antibodies were used: desmin (clone D33), muscle-specific actin (clone HHF35), myogenin (clone F5D), cytokeratin cocktail (clone AE1/AE3), smooth muscle actin (clone 1A4), myosin (clone SMMS-1), CD99 (clone HO36-1.1), CD1a (clone MTB1), alfa fetoprotein (polyclonal), CD45 (clones 2B11 & PD7/26), and epithelial membrane antigen (EMA) (clone E29) at the dilution suggested by the manufacturer. All antibodies were obtained from Cell Marque (Hot Springs, AR, USA).
Conventional Cytogenetic Analysis
A piece of the resected lymph node was minced, cultured in conditioned RPMI 1640 medium supplemented with 25% fetal bovine serum for 72 h. Metaphase chromosomes for Giemsa banding pattern by trypsin digestion with Wright stain (GTW banding) were prepared according to standard procedures. In all, 20 metaphases were karyotyped with CytoVison system (Applied Imaging, Santa Clara, CA, USA), and karyograms were described according to the International System for Human Cytogenetic Nomenclature 2009.22 Coordinates of cells with chromosome aberrations were documented, and the slides were subsequently prepared for metaphase FISH analysis.
Interphase and Metaphase FISH Assays
Touch preparation slides were made from a frozen portion of the lymph node. Interphase FISH was undertaken using LSI FOXO1 (FKHR) dual color break-apart probe at 13q14 (Vysis, Abbott Park, IL, USA) and FGFR1 dual color break-apart probe at 8p12p11.2 (LabCorp, Research Triangle Park, NC, USA). Metaphase FISH was performed on the de-stained GTW slides. Standard FISH protocol was followed. FISH images were captured and analyzed with a CytoVison system (Applied Imaging).
SNP-Based Microarray Analysis
Microarray assay was performed by LabCorp, using Affymetrix (Santa Clara, CA, USA) whole-genome-human SNP array 6.0 platform containing 1.8 million SNP and non-SNP markers. Genomic DNA extraction, probe labeling and hybridization were carried out following the manufacturer's instruction (Affymetrix). The publicly available HapMap set of 270 control individuals and an internal cohort of controls were implemented as control groups. Genotyping Console (GTC) 4.0 software (Affymetrix) was applied to assess copy number alterations and long contiguous stretches of homozygosity (LCSH) using the software default settings. The cut-off for a genomic imbalance was set up as 200 Kb for the deletion and 500 Kb for the duplication. NCBI Build 36.1 (hg18) was used as reference sequence.
Gene Ontology and Data Mining
Gene ontology analysis was conducted using UCSC genome browser (http://genome.ucsc.edu). Functional annotation, pathway hunting, and network construction were performed with Ingenuity Pathway Analysis (IPA) (http://www.ingenuity.com) (Ingenuity, Redwood City, CA, USA). VISTA enhancer browser (http://enhancer.lbl.gov) and ECR browser (http://ecrbase.dcode.org) were used to search for regulatory elements.
Results
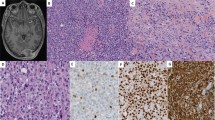
Histopathological study of the lymph node established the diagnosis of metastatic solid variant of alveolar rhabdomyosarcoma. The neoplastic cells had small amounts of lightly eosinophilic cytoplasm and prominent 1–2 centrally located nucleoli (Figure 1a). In some areas, especially toward the periphery, the tumor cells stood like a picket fence at the edge of an empty ‘alveolar’ space. Rare foci of tumor necrosis were present. The tumor cells showed strong diffuse nuclear positive staining for myogenin and focal positive staining for muscle-specific actin (HHF-35) and desmin immunohistochemical stains. The tumor cells also showed membranous and cytoplasmic staining for CD99 and focal membranous and cytoplasmic positivity for EMA and pancytokeratin (Figure 1b-d). They were negative for myosin and CD45. Conventional cytogenetic and FISH studies identified chromosome rearrangements involving FOXO1 and suggested a novel fusion partner. GTW-banding analysis, performed on unstimulated tumor cells, demonstrated an apparently balanced three-way translocation, 46,XX,t(8;13;9)(p11.2;q14;q32), identified as the sole clonal abnormality in 10 of 20 metaphases analyzed (Figure 2a-c). Rearrangement at 13q14 suggested that FOXO1 might be involved. However, loci of the characteristic fusion partners PAX3 at 2q35 and PAX7 at 1p36 seemed not affected, suggesting in this case a novel fusion partner for FOXO1 (Figure 2c). The dual color break-apart FISH probe for FOXO1 used on interphase nuclei to further characterize the cytogenetic aberrations demonstrated one pair of juxtaposed yellow signals for FOXO1 locus, one red signal for the telomeric region of FOXO1, and 10–20 copies of green signals for the centromeric region of FOXO1, indicating that one allele of the FOXO1 gene was rearranged and its centromeric region subsequently amplified (Figure 2d). FISH analysis of de-stained G-banded metaphase cells confirmed translocation of the telomeric region of FOXO1 to the long arm of chromosome 9, and amplification of the centromeric region of FOXO1, with the caveat that the amplification signal could represent a chimerical gene consisting of an unknown DNA sequence at the novel translocation locus 8p11.2 identified by chromosome analysis.

Histopathology and immunohistochemistry of the alveolar rhabdomyosarcoma, solid-variant, metastatic to a lymph node. (a) Sheets of poorly differentiated small round blue tumor cells with prominent nucleoli and scant, pale eosinophilic cytoplasm (hematoxylin–phloxine–saffron staining, × 400). (b) Strong, positive membranous, and cytoplasmic staining for muscle-specific actin (HHF-35, × 400). (c) Strong, positive nuclear staining for myogenin ( × 400) and (d) strong, cytoplasmic and membranous positive staining for desmin ( × 400).

Abnormal cytogenetic findings in the tumor cells. (a) Representative karyogram of a three-way translocation 46,XX,t(8;13;9)(p11.2;q14;q32) observed in 10 of 20 metaphases analyzed. Arrows indicate aberrant chromosomes. (b) Metaphase spread of the same cell karyotyped in panel a. (c) Partial karyograms of seven representative abnormal cells, with no structural changes shown on chromosomes 1 and 2, while chromosomes 8, 9 and 13 display an apparently balanced three-way translocation. Chromosome numbers are listed on the bottom. (d) FISH using dual color break-apart probe for FOXO1 demonstrating one normal fusion signal (yellow), one split signal for the telomeric region of FOXO1 (red), and ∼20 copies of the split signal for the centromeric region of FOXO1 (green). These results indicate a rearrangement at FOXO1 locus, as well as an amplification of its centromeric region.
SNP microarray analysis detected a co-amplification of FOXO1 gene at 13q14 and FGFR1 gene at 8p12-p11. Whole-genome screening for copy number variations using Affymetrix 6.0 microarray platform showed co-amplification of two genomic segments. The first amplification demonstrated approximately 12 copies of a 446-Kb segment (Chr8: 38 304 468–38 750 291) on the short arm of chromosome 8 at p12p11.23 encompassing 293 makers. This segment contains the full-length genomic regions of two reference genes, fibroblast growth factor receptor 1 (FGFR1; NM_023110.2) and leucine zipper-EF-hand containing transmembrane protein 2 (LETM2; NM_001199660.1), and part of the gene Wolf–Hirschhorn syndrome candidate 1-like 1 (WHSC1L1; NM_017778.2) (Figure 3a). The second amplification demonstrated approximately 12 copies of a 255-Kb segment (Chr13: 39 782 030–40 037 118) on the long arm of chromosome 13 at q14.11 encompassing 208 markers. This segment overlaps the centrometric end of forkhead box O1 (FOXO1; NM_002015.3) (Figure 3b). Breakpoints of the above two fragments are consistent with the three-way translocation previously diagnosed by chromosome analysis. In combination with the karyogram and FISH findings, the co-amplification seen on microarray suggested formation of FOXO1–FGFR1 fusion due to chromosome translocation and subsequent amplification of the resultant chimerical product. Using dual color break-apart probe for FGFR1, amplification of the intact locus represented by 10–20 copies of fused yellow signals was observed in all of the interphase nuclei analyzed (Figure 3c). No copy number change was detected for PAX3 or PAX7 by microarray study.

Snapshot of the GTC karyoview and FISH assay depicting copy number alterations detected by affymetric 6.0 SNP microarray analysis. (a) An amplification of 12 copies on the short arm of chromosome 8 at p12-p11.23, spanning approximately 446 Kb. Two full-length reference genes, FGFR1 and LETM2, and one partial gene WHSC1L1 are located inside the amplified area. (b) A second amplification of 12 copies on the long arm of chromosome 13 at q14.11, spanning approximately 255 Kb. This amplified segment overlaps the centromeric region of FOXO1. (c) Amplification of FGFR1 locus was visualized on 100% interphase nuclei analyzed from the tumor by FISH study with dual color break-apart probes, 10–20 fusion signals were seen. Centromeric region of FGFR1 locus was labeled in red and telomeric region of FGFR1 locus was labeled in green, the yellow fusion signal indicates an intact FGFR1 locus. A few split signals represent artifact staining.
In addition, two submicroscopic microdeletions undetectable using current state-of-art cytogenetic techniques were identified by the microarray analysis. The first microdeletion was 2.4 Mb in length (Chr11: 78 493 948–80 917 971), located on the long arm of chromosome 11 at q14.1. The proximal end of this deletion overlaps the promoter and several exons of ODZ4 gene (NM_001098816.2), which is the human homolog of the odd Oz gene in Drosophila (Supplementary Figure 1a). This deletion could presumably disrupt the transcription of ODZ4, but functional information available in the literature is very limited for this gene. Yet, as the major part of this deletion is a non-coding sequence, functional elements regulating gene expression over long distance could reside inside this gene desert. However, no enhancers were found in this area by searching VISTA enhancer browser (http://enhancer.lbl.gov/). Comparative genome analysis was done by aligning this 2.4-Mb human DNA sequence with corresponding regions in rhesus macaque, dog, opossum, rat, mouse, chicken, frog, zebrafish, and fugu genomes (http://ecrbase.dcode.org). In all, 11 fragments were seen evolutionally conserved between human and chicken and one fragment conserved between human and frog, (Supplementary Figure 1b) suggesting that these identified DNA fragments might be non-coding regulator elements. The second finding was a 283-Kb intragenic deletion (ChrX: 32 100 786–32 383 598) across several exons of DMD gene on chromosome X at p21.1, gene responsible for X-linked Duchenne muscular dystrophy. The significance of both deletions in this patient's malignancy is unclear.
The final cytogenetic diagnosis for this patient's tumor is therefore written as 46,XX,t(8;13;9)(p11.2;q14;q32)[10]/46,XX[10].nuc ish amp(FGFR1)[200],(5′FOXO1 × 2, amp(3′FOXO1))(5′FOXO1 con 3′FOXO1 × 1)[90/100]/(FOXO1 × 2)(5′FOXO1 sep 3′FOXO1 × 1)[3/100].arr Xp21.1(32 100 786–32 383 598) × ∼1,8p12p11.23(38 304 468–38 750 291) × ∼12,11q14.1(78 493 948–80 917 971) × 0–1,13q14.11(39 782 030–40 037 118) × ∼12.
Discussion
Histological classification of the tumor and identification of genetic aberrations have significant clinical applications in rhabdomyosarcoma. Although PAX–FOXO1 gene fusion is a characteristic feature for the diagnosis of alveolar rhabdomyosarcoma, translocations involving PAX7 often indicate a better outcome in patients with metastatic disease than that associated with PAX3.4 Yet, 20% of alveolar rhabdomyosarcoma are negative for PAX–FOXO1 rearrangements, and it has remained unclear whether those cases are truly ‘fusion-negative’ or carry chromosomal aberrations at unsuspected novel gene loci,23 as in the case of the t(2;2)(p23;q53) and t(2;8)(q35;q13) described recently, respectively, resulting in fusion genes PAX3-NCOA1 and PAX3-NCOA2.24
PAX proteins are highly specific transcriptional factors crucial for the differentiation of myogenic and neurogenic progenitor cells during early embryonic development.25 In the 80% alveolar rhabdomyosarcoma cases that are PAX–FOXO1 positive, PAX3 and PAX7 DNA-binding domains and FOXO1 transactivation domain are fused. PAX3–FOXO1 or PAX7–FOXO1 chimerical transcripts are overexpressed and activate downstream transcription from PAX protein-binding sites more effectively than their wild-type counterparts. Altered biological behaviors such as dysregulation of angiogenesis, promotion of cell growth, and anti-apoptosis prohibiting differentiation are induced.26, 27 FGFR1 is a tyrosin kinase receptor gene located on the short arm of human chromosome 8 at p12p11. Activating mutations in FGFR4, a family member of FGFR1, were recently detected in 7.5% of rhabdomyosarcoma including both embryonal rhabdomyosarcoma and PAX3–FOXO1-positive alveolar rhabdomyosarcoma tumors.28 Overexpression of FGFR4 was previously found to be associated with advanced stage in rhabdomyosarcoma and particularly alveolar rhabdomyosarcoma.29 Of note, array comparative genomic hybridization (aCGH) on a cohort of 128 primary rhabdomyosarcoma samples showed FGFR1 amplification in 11% PAX–FOXO1-negative alveolar rhabdomyosarcoma cases and 6% embryonal rhabdomyosarcoma cases, but not in alveolar rhabdomyosarcoma patients positive for PAX–FOXO1.19
SNP arrays technology applied to frozen tumor tissue allowed the patient's entire genome to be screened for copy number variation and the amplified fragment, and the unknown FOXO1 fusion partner was characterized. Microarray has been widely used in clinical diagnosis of developmental disorders and in some cases of hematological malignancies, but rarely in solid tumors.30 As opposed to the 5–10-Mb resolution level of conventional chromosome analysis, this array platform allowed detection of submicrospcopic copy number alterations (deletions, duplications, amplifications) and loss of LOH at a submegabase resolution. The results obtained would benefit from additional confirmation, ruling-out unlikely constitutional changes in the peripheral blood.
The 2.4-Mb deletion in 11q14 overlaps the 5′ end of the ODZ4 gene, which is the human homolog of the odd Oz gene in Drosophila with limited functional information in the literature. The majority of this deleted fragment is a gene desert that could harbor regulatory elements (eg enhancers, silencers, insulators, or locus control regions (LCRs)), defects on which could lead to misexpression of the target genes.31 Unfortunately, we were unable to find any experimentally validated enhancer by searching VISTA enhancer browser (http://enhancer.lbl.gov). Although more than 95% of human genome does not code proteins, non-coding regions evolutionarily conserved across vertebrate genomes could suggest regulatory functions.32 When we aligned the 2.4-Mb fragment with the corresponding regions in rhesus macaque, dog, opossum, rat, mouse, chicken, frog, zebrafish, and fugu genomes (http://ecrbase.dcode.org), deeply conserved fragments were seen between human, chicken, and frog (Supplementary Figure 1b). However, the significance of this deletion for this patient's malignancy remains unclear.
Mutations in DMD gene, which were also detected, are at the root cause of the X-linked Duchenne muscular dystrophy.33 Interestingly, DMD gene and other members in the dystrophin-associated glycoprotein (DAG) complex have also been studied in rhabdomyosarcoma but no clear interpretation is available at present, yet it is interesting to note that knockout mice with dystrophin or α-sarcoglycan deficiency spontaneously develop alveolar rhabdomyosarcoma or embryonal rhabdomyosarcoma in aged individuals; however, we could find only two Duchenne muscular dystrophy patients with rhabdomyosarcoma reported in the literature, which may be a coincidence.16, 17
Solid variant of alveolar rhabdomyosarcoma is rare and sometimes mimics other malignancies.34 The 12 cases in the literature since 1994 including the current one are summarized in Supplementary Table 1.34, 39, 40, 41, 42, 43, 44, 45, 46 Fusion gene status was known for 11 patients: eight had PAX3–FOXO1, one had PAX7–FOXO1, and one (the current one) had FGFR1–FOXO1 fusion. Only one patient was negative for identifiable genetic aberration. Myogenin, desmin, vimentin, and actin were all positive for most of the cases.39, 40, 41, 42, 43, 44, 45, 46
Genetic events in rhabdomyosarcoma detected by conventional cytogenetics, FISH, or whole-genome microarray studies affect multiple genes. Manifestation of rhabdomyosarcoma have also been described in several single gene disorders.35 Moreover, signaling pathways such as p53 pathway is tightly involved in the tumorigenesis of rhabdomyosarcoma, as introduction of the fused Pax–Foxo1 is not sufficient to induce the tumor in mice unless other genetic modifications such as p53 mutations were also included.21, 36, 37, 38 Interestingly, online bioinformatic tool IPA revealed interrelationship between most of the collected 31 genes including those genes responsible for developmental disorders (Supplementary Table 1, Supplementary Figure 2).
In summary, using integrated cytogenetic and genomic approaches, we have identified a chromosomal rearrangement that resulted in the formation and amplification of a novel FOXO1–FGFR1 chimerical product in an infant girl with solid variant alveolar rhabdomyosarcoma. Considering the unusual early onset of the tumor, our finding may be a characteristic of a new rhabdomyosarcoma sub-group among the so-called ‘fusion-negative rhabdomyosarcoma’. As opposed to other newly identified translocation loci in alveolar rhabdomyosarcoma such as NCOA1 and NCOA2, FGFR family members have established roles in tumorigenesis. The identification of FGFR1 will help further clarify the molecular mechanisms for rhabdomyosarcoma development, and may also lead to targeted therapies for this type of tumor. We suggest that microarray analysis be performed on all sarcomas with histological evidence of alveolar features, but negative for an identifiable PAX–FOXO1 translocation.
References
Breneman JC, Lyden E, Pappo AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 2003;21:78–84.
Koscielniak E, Harms D, Henze G, et al. Results of treatment for soft tissue sarcoma in childhood and adolescence: a final report of the German Cooperative Soft Tissue Sarcoma Study CWS-86. J Clin Oncol 1999;17:3706–3719.
Parham DM . Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod Pathol 2001;14:506–514.
Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children's oncology group. J Clin Oncol 2002;20:2672–2679.
Sawyer JR, Crussi FG, Kletzel M . Pericentric inversion (2)(p15q35) in an alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 1994;78:214–218.
Anderson J, Gordon A, Pritchard-Jones K, et al. Genes, chromosomes, and rhabdomyosarcoma. Genes Chromosomes Cancer 1999;26:275–285.
Prezioso D, Lotti T, Montironi R, et al. Role of neoadjuvant treatment in clinically confined prostate cancer. Takeda NHT Italian Group. Eur Urol 1999;35 (Suppl 1):17-–21; discussion 2.
Bridge JA, Liu J, Weibolt V, et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: an intergroup rhabdomyosarcoma study. Genes Chromosomes Cancer 2000;27:337–344.
Loh Jr WE, Scrable HJ, Livanos E, et al. Human chromosome 11 contains two different growth suppressor genes for embryonal rhabdomyosarcoma. Proc Natl Acad Sci USA 1992;89:1755–1759.
Schaaf GJ, Ruijter JM, van Ruissen F, et al. Full transcriptome analysis of rhabdomyosarcoma, normal, and fetal skeletal muscle: statistical comparison of multiple SAGE libraries. FASEB J 2005;19:404–406.
Forus A, Florenes VA, Maelandsmo GM, et al. Mapping of amplification units in the q13-14 region of chromosome 12 in human sarcomas: some amplica do not include MDM2. Cell Growth Differ 1993;4:1065–1070.
Khatib ZA, Matsushime H, Valentine M, et al. Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res 1993;53:5535–5541.
Roberts WM, Douglass EC, Peiper SC, et al. Amplification of the gli gene in childhood sarcomas. Cancer Res 1989;49:5407–5413.
Shapiro DN, Jones BG, Shapiro LH, et al. Antisense-mediated reduction in insulin-like growth factor-I receptor expression suppresses the malignant phenotype of a human alveolar rhabdomyosarcoma. J Clin Invest 1994;94:1235–1242.
Jones AE, Albano EA, Lovell MA, et al. Metastatic alveolar rhabdomyosarcoma in multiple endocrine neoplasia type 2A. Pediatr Blood Cancer 2010;55:1213–1216.
Jakab Z, Szegedi I, Balogh E, et al. Duchenne muscular dystrophy-rhabdomyosarcoma, ichthyosis vulgaris/acute monoblastic leukemia: association of rare genetic disorders and childhood malignant diseases. Med Pediatr Oncol 2002;39:66–68.
Rossbach HC, Lacson A, Grana NH, et al. Duchenne muscular dystrophy and concomitant metastatic alveolar rhabdomyosarcoma. J Pediatr Hematol Oncol 1999;21:528–530.
Anderson JR, Barr FG, Hawkins DS, et al. Fusion-negative alveolar rhabdomyosarcoma: modification of risk stratification is premature. J Clin Oncol 2010;28:e587–e588; author reply e9-90.
Williamson D, Missiaglia E, de Reynies A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol 2010;28:2151–2158.
Wexler LH, Ladanyi M . Diagnosing alveolar rhabdomyosarcoma: morphology must be coupled with fusion confirmation. J Clin Oncol 2010;28:2126–2128.
Matsumura T, Yamaguchi T, Seki K, et al. Advantage of FISH analysis using FKHR probes for an adjunct to diagnosis of rhabdomyosarcomas. Virchows Arch 2008;452:251–258.
Shaffer LG, Slovak ML, Campbell LJ (eds). ISCN (2009): International System of Human Cytogenetic. Nomenclature S Karger AG, Basel, 2009.
Mercado GE, Barr FG . Fusions involving PAX and FOX genes in the molecular pathogenesis of alveolar rhabdomyosarcoma: recent advances. Curr Mol Med 2007;7:47–61.
Sumegi J, Streblow R, Frayer RW, et al. Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Cancer Genet Cytogenet 2010;49:224–236.
Chi N, Epstein JA . Getting your Pax straight: Pax proteins in development and disease. Trends Genet 2002;18:41–47.
Barr FG . Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001;20:5736–5746.
Helman LJ, Meltzer P . Mechanisms of sarcoma development. Nat Rev Cancer 2003;3:685–694.
Taylor VI JG, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest 2009;119:3395–3407.
Davicioni E, Finckenstein FG, Shahbazian V, et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 2006;66:6936–6946.
Heinrichs S, Li C, Look AT . SNP array analysis in hematologic malignancies: avoiding false discoveries. Blood 2010;115:4157–4161.
Nobrega MA, Zhu Y, Plajzer-Frick I, et al. Megabase deletions of gene deserts result in viable mice. Nature 2004;431:988–993.
Loots G, Ovcharenko I . ECRbase: database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes. Bioinformatics 2007;23:122–124.
Hoffman EP, Brown Jr RH, Kunkel LM . Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919–928.
Ganesan P, Thulkar S, Rajan A, et al. Solid variant of alveolar rhabdomyosarcoma mimicking non-Hodgkin lymphoma: case report and review of literature. J Pediatr Hematol Oncol 2008;30:772–774.
Jones AE, Albano EA, Lovell MA, et al. Metastatic alveolar rhabdomyosarcoma in multiple endocrine neoplasia type 2A. Pediatr Blood Cancer 2010;55:1213–1216.
Keller C, Arenkiel BR, Coffin CM, et al. Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 2004;18:2614–2626.
Gordon AT, Brinkschmidt C, Anderson J, et al. A novel and consistent amplicon at 13q31 associated with alveolar rhabdomyosarcoma. Genes Chromosomes Cancer 2000;28:220–226.
Gang EJ, Bosnakovski D, Simsek T, et al. Pax3 activation promotes the differentiation of mesenchymal stem cells toward the myogenic lineage. Exp Cell Res 2008;314:1721–1733.
Parham DM, Shapiro DN, Downing JR, et al. Solid alveolar rhabdomyosarcomas with the t(2;13). Report of two cases with diagnostic implications. Am J Surg Pathol 1994;18:474–478.
Bianchi L, Orlandi A, Iraci S, et al. Solid alveolar rhabdomyosarcoma of the hand in adolescence: a clinical, histologic, immunologic, and ultrastructural study. Pediatr Dermatol 1995;12:343–347.
Yule SM, Bown N, Malcolm AJ, et al. Solid alveolar rhabdomyosarcoma with a t(2;13). Cancer Genet Cytogenet 1995;80:107–109.
Sartelet H, Lantuejoul S, Armari-Alla C, et al. Solid alveolar rhabdomyosarcoma of the thorax in a child. Histopathology 1998;32:165–171.
Smith A, Sharma P, Tomlinson J, et al. Solid variant of alveolar rhabdomyosarcoma with unbalanced t(2;13) and hypotetraploidy, without MYCN amplification. Pathology 2001;33:108–111.
Senger C, Diaz L, Katzenstein H, et al. Pathologic quiz case: ovarian mass in a 2-year-old girl presenting with pleural effusions. Arch Pathol Lab Med 2003;127:e56–e59.
Cerveira N, Torres L, Ribeiro FR, et al. Multimodal genetic diagnosis of solid variant alveolar rhabdomyosarcoma. Cancer Genet Cytogenet 2005;163:138–143.
Passmore LM, Myers P, Gilbert-Barness E . Pathology teach and tell: solid variant alveolar rhabdomyosarcoma of the orbit. Fetal Pediatr Pathol 2006;25:51–57.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Liu, J., Guzman, M., Pezanowski, D. et al. FOXO1–FGFR1 fusion and amplification in a solid variant of alveolar rhabdomyosarcoma. Mod Pathol 24, 1327–1335 (2011). https://doi.org/10.1038/modpathol.2011.98
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.98
Keywords
This article is cited by
-
Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
Molecular Cancer (2024)
-
Biological and clinical implications of FGFR aberrations in paediatric and young adult cancers
Oncogene (2023)
-
Re-evaluating tumors of purported specialized prostatic stromal origin reveals molecular heterogeneity, including non-recurring gene fusions characteristic of uterine and soft tissue sarcoma subtypes
Modern Pathology (2021)
-
Soft Tissue Special Issue: Skeletal Muscle Tumors: A Clinicopathological Review
Head and Neck Pathology (2020)
-
The current landscape of rhabdomyosarcomas: an update
Virchows Archiv (2020)
