
Abstract
Fibroblast growth factor receptor 2 (FGFR2) is a tyrosine kinase receptor involved in many biological processes such as embryogenesis, adult tissue homeostasis and cell proliferation. Mutations in FGFR2 have been reported in up to 10–12% of endometrial carcinomas identical to those found in congenital craniofacial disorders. Inhibition of FGFR2 could be a new therapeutic target in endometrial carcinoma. FGFR2 immunostaining was assessed in three tissue microarrays: one constructed from paraffin-embedded blocks of 60 samples of normal endometrium in different phases of menstrual cycle, and two tissue microarrays containing endometrial carcinoma samples (95 and 62 cases). FGFR2 expression was correlated with stage, histological type and grade as well as with immunostaining of PTEN, RASSF1A, estrogen and progesterone receptors, KI67, Cyclin D1, STAT-3 and SPRY2. FGFR2 mutations were assessed by PCR and direct sequencing, with DNA obtained from 31 paraffin-embedded endometrial carcinoma samples. In normal endometrium, FGFR2 expression was higher in the secretory than in the proliferative phase (P=0.001), with an inverse correlation with Ki67 (P=0.00032), suggesting a tumor-suppressor role for FGFR2 in normal endometrium. Cytoplasmic expression of FGFR2 was higher in endometrial carcinoma when compared with the atrophic endometrium from the same patients (P=0.0283), but was lower in comparison with normal endometrium from women in the menstrual cycle. Interestingly, nuclear staining was observed in some cases, and it was less frequent in endometrial carcinoma when compared with the adjacent atrophic endometrium (P=0.0465). There were no statistical differences when comparing superficial and myoinvasive endometrial carcinoma samples. Endometrioid endometrial carcinomas showed higher expression of FGFR2 than nonendometrioid endometrial carcinomas (fold change 2.56; P=0.0015). Grade III endometrioid endometrial carcinomas showed decreased FGFR2 expression when compared with grade II endometrioid endometrial carcinomas (P=0.0055). No differences were found regarding pathological stage. Two missense mutations of FGFR2 gene were detected in exons 6 and 11 (S252W and N549K, respectively; 6.45%). Results support the hypothesis that FGFR2 has a dual role in the endometrium, by inhibiting cell proliferation in normal endometrium during the menstrual cycle, but acting as an oncogene in endometrial carcinoma.
Similar content being viewed by others


Main
Endometrial carcinoma is one the most commonly diagnosed malignant tumors of the female genital tract in Europe and United States.1 There are two main clinicopathological variants of endometrial carcinoma. Endometrioid carcinomas (type I tumors)2 are usually low-grade and estrogen-related carcinomas that usually develop in perimenopausal women and coexist or are preceded by complex and atypical endometrial hyperplasia. Nonendometrioid carcinomas (type II tumors) are very aggressive tumors, unrelated to estrogen stimulation, arising occasionally in endometrial polyps or from precancerous lesions developing in atrophic endometrium that mainly occur in older women; and may exhibit serous or clear cell features. Classification of endometrial carcinomas in these two types is probably too rigid, as tumors showing combined or mixed features are not infrequent in daily practice. However, cDNA analysis clearly has shown that endometrioid endometrial carcinomas and nonendometrioid endometrial carcinomas exhibit different expression profiles.3 The molecular alterations involved in the development of endometrioid endometrial carcinomas are different from those of nonendometrioid endometrial carcinomas. Nonendometrioid endometrial carcinomas exhibit alterations of p53,4 STK15,3 p16, E-cadherin,5 and C-erbB2, as well as loss of heterozygosity on several chromosomes.6 In contrast, endometrioid endometrial carcinomas show microsatellite instability,7 and mutations in the PTEN,8 KRAS,9 and CTNNB-1 genes.10, 11, 12
Oncogenic activation of tyrosine kinases is a common mechanism of carcinogenesis. Tyrosine kinases play a role in transduction of proliferating signals and can be good therapeutic target in several tumors. Recently, some investigators have suggested that alterations in fibroblast growth factor receptor 2 (FGFR2) could be added to the molecular-specific features of endometrioid endometrial carcinomas.13, 14, 15 FGFR2 is of special interest, as it is a possible target for therapeutic approaches, and FGFR2 inhibitors are currently under consideration in clinical trials for several types of solid tumors. FGFR2 belongs to fibroblast growth factor receptor (FGFR) tyrosine kinase family that comprises four different transmembrane kinases (FGFR1–FGFR4) and their alternative spliced isoforms.16 They differentially respond to 18 FGF ligands and activate downstream pathways such as RAS-MAPK. FGFR2 has been described to play a role as either oncogene or tumor-suppressor gene, depending on the type of cell. Several types of molecular alterations have been described, including gene overexpression and point mutation. In this study, we evaluate FGFR2 expression in normal endometrium and endometrial carcinoma by immunohistochemistry and the presence of somatic mutations of FGFR2 by PCR in correlation with the main molecular alterations of this tumor.
Materials and methods
Material
Tissue samples were obtained from Hospital Universitari Arnau de Vilanova de Lleida and Hospital de Sant Pau, Barcelona. A specific informed consent was obtained from each patient, and the study was approved by the local ethical committee. The material included 60 samples of normal endometrial tissue that were fixed in formalin and embedded in paraffin (20 proliferative, 40 secretory). A total of 157 samples corresponded to endometrial carcinomas: 131 of them were endometrioid endometrial carcinomas and the other 26 were nonendometrioid endometrial carcinomas. Formalin-fixed, paraffin-embedded blocks were available for each of them. Overall, the series of 157 endometrial carcinoma included 47 endometrioid endometrial carcinomas grade I, 58 endometrioid endometrial carcinomas grade II, 26 endometrioid endometrial carcinomas grade III, 15 serous carcinomas, 4 clear cell carcinomas and 7 mixed müllerian malignant tumors. In all, 108 tumors were stage I, 15 were stage II, 22 were stage III and 1 was stage IV. Staging information was incomplete in 11 cases. Furthermore, superficial and deep tumor samples of the same patient were taken in 34 cases.
Tissue Microarrays
Three tissue microarrays were designed. The first tissue microarray was constructed from 60 paraffin-embedded samples on normal endometrium in different phases of menstrual cycle (20 proliferative, 40 secretory). The second tissue microarray was composed of 95 endometrial carcinomas, previously evaluated for microsatellite instability, KRAS mutations and alterations in PTEN, PIk3CA and CTNNB-1.7, 8, 9, 10, 11, 17 The third tissue microarray was constructed from 62 endometrial carcinomas that were also previously subjected to molecular analysis for genes involved in the control of RAS-MAPK pathway, such as SPRY2, and RASSF1A promoter hypermethylation.18, 19 A Tissue Arrayer device (Beecher Instruments, Sun Prairie, WI, USA) was used to construct the tissue microarray. Briefly, all the samples were histologically reviewed and representative areas were marked in the corresponding paraffin blocks. Two selected cylinders (0.6 mm in largest diameter) from two different areas were included in each case. Control normal tissues from the same endometrial carcinoma specimens were also included.
Immunohistochemical Study
Tissue microarray blocks were sectioned at a thickness of 3 μm, dried for 16 h at 56°C before being dewaxed in xylene and rehydrated through a graded ethanol series, and washed with phosphate-buffered saline. Antigen retrieval was achieved by heat treatment in a pressure cooker for 2 min in EDTA (pH 8.9). Before staining the sections, endogenous peroxidase was blocked. The antibodies used were: FGFR2 (1:200 dilution; Abcam), SPRY2 (1:500 dilution; N-terminal, S 1444, Sigma), Ki67 (1:100 dilution; MIB-1, DAKO), PTEN (1:50 dilution; 6H2.1, DAKO), RASSF1A (1:100 dilution; eB114-10H1; e-Bioscience), Estrogen receptor (1:50, 6F11, NovoCastra), Progesterone receptor (1:50, PgR 636, DAKO), Cyclin D1 (1:25, DCS6, DAKO) and STAT-3 (1:500, sc8019, Santa Cruz). After incubation, the reaction was visualized with the EnVision Detection Kit (DAKO) using diaminobenzidine chromogen as a substrate. Sections were counterstained with hematoxylin. Appropriate positive and negative controls were also tested.
Immunohistochemical results were evaluated by two pathologists, by following uniform pre-established criteria. FGFR2 immunoexpression, as well as staining for the other markers, was graded semiquantitatively by considering the percentage and intensity of the staining. A histological score was obtained from each sample, which ranged from 0 (no immunoreaction) to 300 (maximum immunoreactivity). The score was obtained by applying the following formula: Histoscore=1 × (% light staining) + 2 × (% moderate staining) + 3 × (% strong staining). The reliability of such score for interpretation of immunohistochemical staining in endometrial carcinoma tissue microarrays has been shown previously.20, 21, 22 As each tissue microarray included two different tumor cylinders from each case, immunohistochemical evaluation was done after examining both samples. Finally, the percentage of positive nuclei in each case was used to assess the cellular proliferation (Ki67).
The reproducibility of tissue microarray immunostaining was confirmed by comparing tissue microarray's results with those obtained in sections from the corresponding paraffin blocks of 37 randomly selected cases. The overall concordance was 89.2%. The κ index of agreement between the two methods ranged from 0.68 to 0.83.
Mutation Analysis of FGFR2
For PCR, two primer pairs were used to individually amplify 12 exons of FGFR2 from genomic endometrial cancer DNA. Each PCR reaction contained 1 × Buffer, 1.5 mM MgCl2 and 1u/μl TaqGold (Applied Biosystems, Santa Clara, CA, USA), 0.2 mM dNTPs (Biotools B&M Labs, SA, Madrid, Spain) and 0.2 μM of each primer. PCR amplification was performed in 20 μl reaction volumes that contained 100 ng of DNA, 75 nM HCl, 1.5 mM MgCl2, 50 mM KCl, 20 mM (NH4)2SO4, 0.2 μM of each primer, 0.2 mM of each dNTP and 1 unit of Taq DNA polymerase (Biotools, B&M Labs). All exons were amplified with the following conditions: an initial 5-min denaturation at 94 °C followed by 35 cycles of 1 min at 94 °C, 1 min at 57 °C and a final extension of 10 min at 72 °C.
For DNA sequencing, PCR products were first purified using the MinElute PCR Purification Kit (Qiagen GmbH, Hilden, Germany) and were bidirectionally sequenced using the original primer pair and the Applied Biosystem Cycle Sequencing kit (Applied Biosystems). Samples were analyzed on the AB Prism 3100-Avant instrument, using standard run parameters. The separation matrix used was POP-6 using 1 × TBE with EDTA running buffer (Applied Biosystems).
Statistical Analysis
To evaluate differences in immunoexpression for independent series, Mann–Whitney nonparametric test was used to evaluate significance. Histoscore means were computed to report absolute difference and, in addition, fold change was calculated to report relative differences. Regarding dependent series, where normal and tumor samples were obtained from the same patients, linear mixed models were used to take into account both paired observations and repeated measures, using model estimates to evaluate differences in expression levels. To evaluate coexpression between biomarkers, Pearson's linear correlation coefficient was applied whereas Spearman's nonparametric test was used to assess significance, which was set for all tests at a threshold of 0.05. All analyses were obtained using R statistical software.
Results
FGFR2 Expression by Immunohistochemical Analysis of Tissue Microarrays
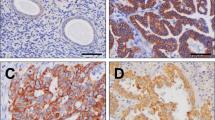
In normal endometrial tissue, FGFR2 protein expression was evaluated in 30 samples, which corresponded to different phases of the menstrual cycle (Table 1 and Figure 1). The remaining samples were missed in the construction of the tissue microarray, lost during sectioning and staining, or did not show representative endometrial glands in the stained section. FGFR2 expression was variable in epithelial cells with some staining in stromal cells and capillaries. Cytoplasmic FGFR2 immunostaining was significantly lower in the proliferative phase (mean Hscore 64; range 0–150) in comparison with the secretory phase (mean Hscore 170; range 10–275) (fold change=2.65; P=0.001; Figure 2). The staining was predominantly cytoplasmic and granular. FGFR2 protein expression was inversely correlated with estrogen (mean Hscore 198; range 0–300) and progesterone (mean Hscore 160; range 0–300) receptors (r=−0.76; P=0.00002; and r=−0.75; P=0.00001, respectively) and was also inversely correlated with Ki-67 staining (mean percentage 14; range 0–70; r=−0.35; P=0.00032; Table 2).

(a) Differential immunoexpression between secretory and proliferative samples. Bars represent means and segments means±1 s.d. Fold change indicates the relative difference between sample types. (b) Differential immunoexpression between normal and tumor samples. Bars represent means and segments means±1 s.d. Fold change indicates the relative difference between sample types.

Cytoplasmic FGFR2 expression in normal endometrium in the proliferative (a) and secretory (b) phases.
In endometrial carcinomas, FGFR2 was evaluated in 115 out of the 157 cases that were included in the second and the third tissue microarrays. The remaining samples were missed in the construction of the tissue microarray, lost during sectioning and staining, did not show representative tumor in the stained section, or had unreliable tissue immunostaining. FGFR2 showed two different types of staining patterns: cytoplasmic and nuclear. Cytoplasmic FGFR2 expression was higher in endometrial carcinomas in comparison with normal atrophic endometrial tissue from the same patient, which was available in 72 cases (fold change 3.35; P=0.0283; Figure 3 and Table 3). However, cytoplasmic FGFR2 expression of endometrial carcinomas was lower than FGFR2 expression in normal endometrium from samples of the first tissue microarray, obtained from premenopausal patients in different phases of the menstrual cycle. Differences in the expression were significant for all normal endometrium samples (fold change 1.47; P=0.013) and for normal endometrium samples in the secretory phase (fold change 1.86; P=0.00016), but not for normal endometrium samples in the proliferative phase (P=0.34). Interestingly, nuclear staining was observed in some cases, and it was less frequent in endometrial carcinomas when compared with atrophic endometrium of the same patients (P=0.0465; Figure 4). In some cases, nuclear staining was seen in atrophic glands with tubal metaplasia. Nuclear staining was not seen in normal endometrium samples in different phases of the menstrual cycle.

Cytoplasmic FGFR2 expression in endometrial carcinoma (a) and the adjacent atrophic endometrium (b) of the same patient.

Nuclear FGFR2 expression is lower in endometrial carcinoma (a) than the adjacent atrophic endometrium (b) of the same patient.
Interestingly, the relationship between FGFR2 expression and estrogen and progesterone expression in endometrial carcinomas was different from the one obtained in normal endometrium during the menstrual phase. In endometrial carcinoma, FGFR2 was significantly associated with increased expression of estrogen receptors (mean Hscore 67; range 0–280; r=0.44; P=0.00002) and progesterone receptors (mean Hscore 96; range 0–290; r=0.22; P=0.00122). Moreover, FGFR2 expression was statistically inversely associated with PTEN immunostaining (mean Hscore 40.52; range 0–290; r=−0.35; P=0.00005). A significant association was also seen when correlating the expression of FGFR2 and SPRY2 (r=0.22; P=0.014). In addition, FGFR2 expression did not show any statistical association with the expression of Ki-67, RASSF1A, STAT-3 and Cyclin D1 (Table 2). Moreover, FGFR2 expression did not show statistical association with KRAS mutations, in the 34 tumors in which KRAS mutation status was known (P=0.85).
Among endometrial carcinoma, FGFR2 immunostaining was higher in endometrioid endometrial carcinoma (mean Hscore 98; range 0–275) in comparison with the nonendometrioid endometrial carcinoma (mean Hscore 38; range 0–260), and the difference was statistically significant (fold change 2.56, P=0.0015; Figure 5). There were no statistical differences when comparing superficial and myoinvasive tumor samples (P=0.5146). Regarding histological grade, no significant differences were seen between grade I and grade II endometrial carcinomas. However, grade III endometrial carcinomas (mean Hscore 54.57; range 0–130) showed decreased FGFR2 expression when compared with grade II endometrial carcinomas (mean Hscore 118.01; range 0–275; fold change=2.16; P=0.0055). No differences were found regarding pathological stage.

Cytoplasmic FGFR2 expression is higher in endometrioid carcinomas of the endometrium (a) in comparison with nonendometrioid carcinomas (b).
FGFR2 Mutations
Missense mutations of FGFR2 gene were detected in exons 6 and 11 (S252W and N549K, respectively) in two of the 31 assessed cases (6.45%), each of them in endometrioid endometrial carcinoma (Figure 6). None of the remaining 29 cases exhibited any mutation. These 31 cases had been previously evaluated for mutations in PTEN (15 of 31; 48%), KRAS (7 of 30; 23%), CTNNB1 (5 of 29; 17%), PIK3CA (9 of 31; 29%) and also for microsatellite instability (13 of 31, 41%).7, 8, 9, 10, 17 The tumor that exhibited the N549K mutation also had one mutation in PTEN (993delC) and one mutation in PIK3CA (H1065L), whereas the S252W FGFR2 mutation coexisted with two mutations in PIK3CA (T1052K and E542V) and one mutation in CTNNB-1. None of these two cases showed a mutation in KRAS. DNA polymorphisms in FGFR2 were very frequent (Figure 7), particularly the V232V change. DNA polymorphisms in exon 5 were seen in 85% of the cases, and in 50% were detected in homozygosis.

FGFR2 mutations in endometrioid carcinomas: S252W (a) and N549K (b).

FGFR2 DNA polymorphism in exon 5.
Discussion
FGFs comprise a large group of heparin-binding growth factors that include 18 ligands. FGFs are expressed in almost all tissues, and play important roles in normal and neoplastic cells by regulating development, wound repair and angiogenesis. FGFs exert their function through four high-affinity tyrosine kinase receptors (FGFR1, FGFR2, FGFR3 and FGFR4).16 Each receptor contains three Ig-like extracellular domains, a transmembrane region and intracellular domain with tyrosine kinase activity and a carboxy terminus. Binding of FGFs to the extracellular domains of FGF receptor results in dimerization and conformational shift in receptor structure that leads to activation of the intracellular kinase domain and subsequent transphosphorylation of tyrosine kinase domains. Activation of FGF receptors leads to signal transduction through multiple pathways including PLCγ, PI3K, MAPK and STATs. However, one of the predominant signaling pathway activated downstream of FGFR is RAS-MAPK. Following FGFR activation, MAPK signaling induces cell proliferation, through induction of Cyclin D1. FGF signaling is under the regulatory control of some proteins. Among them, the members of the Sprouty mammalian genes (SPRY)23 seem to have a significant role. The mode of action of SPRY proteins is complex and subjected to several unknown mechanisms of regulation. SPRY proteins have been found to regulate both receptor tyrosine kinases (RTKs) and downstream signaling pathways like FGF and RAS-MAPK.24 By controlling these pathways, SPRY2 is involved in regulation of cell proliferation, differentiation and angiogenesis.
FGF signaling can be involved in tumor development or progression by different mechanisms in a context-dependent manner. In some scenarios, FGF activation may have oncogenic roles by increasing cell proliferation, survival and migration. However, in other settings, FGF signaling may also play tumor-suppressor roles.25 In fact, loss of FGFR2 signaling may induce epithelial-to-mesenchymal transition26 in tumor cells. Overexpression of FGFRs has been identified in tumors from the brain, thyroid, breast, prostate and skin. Copy number gains of FGFR2 have been shown to result in FGFR2 overexpression in cancers from the breast27 and stomach. Moreover, missense mutations of some of the FGFR have been reported. FGFR2 mutations have been described in cancers from the ovaries, breast, stomach28 and lung.29 FGFR2 mutations around the third immunoglobulin-like domain result in FGFR2 activation due to the creation of autocrine FGF signaling loop. Moreover, mutations involving the tyrosine kinase domain cause FGFR2 activation by release of FGFR2 from autoinhibition. The important role of FGF signaling in neoplastic development can be suggested after demonstration of high prevalence of single-nucleotide polymorphisms (SNPs)30 within intron 2 of FGFR2 in association with breast cancer through allelic FGFR2 upregulation.
As mentioned before, the RAS-MAPK is an important signaling pathway downstream of FGFR. Activation of the pathway begins when a signal binds to a protein kinase receptor such as FGFR, and also EGFR or PDGFR, although it may also be activated by many other multiple upstream receptors. Deregulation of the RAS signaling pathway plays an important role in endometrial carcinoma.9 The frequency of KRAS mutations in endometrial carcinoma ranges between 10 and 30%. In some series, KRAS mutations have been reported to be more frequent in endometrioid endometrial carcinomas showing microsatellite instability. Inactivation of RASSF-1A, which is a negative regulator of the RAS-MAPK pathway, is also frequent in endometrial carcinoma. In a recent study, we found decreased expression of RASSF1A18 in almost 50% of the cases. Reduced expression of RASSF1A was frequently associated with RASSF1A promoter hypermethylation.
There are several evidences suggesting that the FGF signaling pathway is important in endometrial carcinoma. Recent studies have shown that endometrial carcinoma presents frequent inactivation of SPRY2, a protein that is involved in the negative regulation of the FGFR pathway. The expression of SPRY2 has been found to be decreased in several types of human cancer, by mechanisms of promoter methylation. In a recent study, we have found reduced SPRY2 immunoexpression in almost 20% of endometrial carcinoma, which was strongly associated with increased cell proliferation.19 In this very same series of cases, SPRY2 promoter methylation was detected in >50% of the cases. Moreover, somatic mutations in the receptor tyrosine kinase FGFR2, identical to the germline mutations associated with craniosynostosis and skeletal dysplasia syndromes, have been recently detected in 10–12% of endometrial carcinomas, particularly in endometriod endometrial carcinomas (16%).13, 14, 15 The somatic mutations included the S252W and P235R changes, which are associated with the Apert syndrome, the N549K and K659M, which are associated with Crouzon syndrome, as well as the N550K change.31 Interestingly, FGFR2 and K-RAS mutations were mutually exclusive events, whereas mutations in FGFR2 and PTEN frequently coexisted. Ectopic expression of one FGFR2 mutation (S252W) in NIH 3T3 cells conferred anchorage-independent growth, suggesting an oncogenic role for FGFR2 mutations. Moreover, downregulation of FGFR2 induced cell cycle arrest and cell death, independently to the status of PTEN.15
In the present study, we have assessed the role of FGFR2 in endometrial carcinomas. First, we have evaluated FGFR2 expression in normal endometrium, by correlating FGFR2 immunohistochemical staining between samples in different phases of the menstrual cycle. Then, we compared FGFR2 expression between endometrial carcinoma and the normal endometrium samples obtained in patients in different phases of the menstrual cycle, and also with samples corresponding to the atrophic endometrium adjacent to the neoplastic tissue, of the same patients with endometrial carcinoma. The results were very interesting. In normal endometrium during the menstrual cycle, FGFR2 seems to be associated with decreased proliferative activity, as the expression is higher in the secretory endometrium, and FGFR2 staining showed an inverse statistical association with expression of estrogen and progesterone receptors and Ki-67 (MIB-1). In other words, in normal endometrium, FGFR2 seems to play an inhibitory (tumor suppressor) function, different to the oncogenic role that has been shown to present in endometrial carcinoma. We have mentioned before that FGFR2 has shown to exhibit this dual activity, by either promoting or inhibiting cell growth, in different cell types in distinct contexts. Now, we show that these different roles may take place in the endometrium, as FGFR2 expression is associated with decreased proliferation in normal endometrium. It is important to remember that other genes also exhibit different roles in normal and neoplastic tissues. It is worth mentioning that TGFβ acts as a tumor suppressor in normal tissue, but plays an oncogenic role in tumors,32 by promoting cell growth and development of epithelial-to-mesenchymal transition.33, 34 Interestingly, in our study, we identified two different patterns of FGFR2 immunostaining: cytoplasmic and nuclear. Although cytoplasmic expression was higher in endometrial carcinoma in comparison with the adjacent atrophic endometrium from the same patients, nuclear FGFR2 expression was higher in the atrophic endometrium. These results suggest that a shift of FGFR2 from the cytoplasm to the nucleus may be associated with decreased oncogenic activity. There have been reports that FGFR2 may localize to the nucleus in various cell types. For example, nuclear expression of FGFR2 has been observed in Sertoli cell precursors upon FGF9 activation.35
As expected, FGFR2 expression was higher in endometrioid endometrial carcinoma than in nonendometrioid endometrial carcinoma, and the difference had statistical significance. In endometrial carcinoma, FGFR2 immunostaining was statistically significantly associated with estrogen and progesterone receptors and inversely associated with PTEN expression, whereas there was not an inverse correlation with SPRY2 expression (an inhibitor of FGF signaling),23 and there was not significant association with the expression of three proteins related to signaling pathways downstream of FGFR2, such as RASSF-1A, Cyclin D1 (RAS-MAPK) and STAT-3. These results were unexpected. We would expect an inverse relationship of FGFR2 with SPRY2 and RASSF1A, as these are negative regulators of FGF and RAS-MAPK signaling pathways, while we would also expect a good correlation between FGFR2 expression and the immunohistochemical staining of STAT-3 and Cyclin D1, which are proteins activated as a result of signaling pathways downstream FGFR2. The lack of correlation between these proteins probably reflects that many different signaling pathways are interconnected, following diverse genetic and epigenetic alterations, and emphasizes the difficulties of detecting in tumor tissue good correlations between molecular alterations and the expected targets. It is worth mentioning that increased Cyclin D1 staining may also occur in endometrial carcinoma with microsatellite instability, in association with specific mutations in Cyclin D1, which prevent Cyclin D1 degradation,36 and such a phenomenon is independent of RAS-MAPK activation.
Moreover, when correlating FGFR2 staining and pathological features, we saw that grade III endometrioid endometrial carcinoma showed decreased FGFR2 expression when compared with grade II endometrioid endometrial carcinoma (P=0.0055). In addition, no differences were found in FGFR2 expression regarding pathological stage, and there were no statistical differences when comparing tumor cells in the superficial part and those in the front of myometrial invasion.
Mutation analysis of FGFR2 demonstrated the presence of missense mutations in 2 of the 31 assessed cases. These two cases were endometrioid endometrial carcinoma. They were detected in exons 6 and 11 (S252W and N549K, respectively). Overall, FGFR2 mutations were detected in 6.45% of endometrial carcinomas, whereas none of the nonendometrioid endometrial carcinoma cases exhibited any mutation. The frequency of FGFR2 mutations in this series (6.45%) is a little bit lower than that of previous series (10–12%).13, 14, 15 However, the number of cases tested for FGFR2 mutations is smaller than that of other series. It is worth mentioning that our series has the additional value that the cases had been previously tested for microsatellite instability, as well as mutations of KRAS, PTEN, PIK3CA and CTNNB1. As shown in other series, none of the tumors that presented a FGFR2 mutation had KRAS mutations, whereas FGFR2 mutations coexisted with mutations in PTEN, PIK3CA and CTNNB-1. Interestingly, we identified many DNA polymorphisms in the coding sequence of FGFR2 in our series of cases.
In summary, in our study we provide additional information regarding the oncogenic role of FGFR2 in endometrial carcinoma, and we also show that FGFR2 has a growth-inhibitory function in normal endometrium. Our results give support to the hypothesis that FGFR2 may be a good target for therapeutic intervention in endometrial carcinoma.
References
Bockman JV . Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983;15:10–17.
Matias-Guiu X, Catasus L, Bussaglia E, et al. Molecular pathology of endometrial hyperplasia and carcinoma. Hum Pathol 2001;32:569–577.
Moreno-Bueno G, Sánchez-Estévez C, Cassia R, et al., Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: STK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res 2003;63:5697–5702.
Prat J, Oliva E, Lerma E, et al., Uterine papillary serous adenocarcinoma. A 10 case study of p53 and c-erbB 2 expression and DNA content. Cancer 1994;74:1778–1783.
Moreno-Bueno G, Hardisson D, Sarrió D, et al., Abnormalities of E- and P-cadherin and catenin (beta-, gamma-catenin, and p120ctn) expression in endometrial cancer and endometrial atypical hyperplasia. J Pathol 2003;199:471–478.
Velasco A, Pallares J, Santacana M, et al. Loss of heterozygosity in endometrial carcinoma. Int J Gynecol Pathol 2008;27:305–317.
Catasús Ll, Machín P, Matias-Guiu X, et al., Microsatellite instability in endometrial carcinomas. Clinico-pathologic correlations in a series of 42 cases. Hum Pathol 1998;29:1160–1164.
Bussaglia E, del Rio E, Matias-Guiu X, et al., PTEN mutations in endometrial carcinomas. A molecular and clinicopathologic analysis of 38 cases. Hum Pathol 2000;31:312–317.
Lagarda H, Catasus Ll, Argüelles RM, et al., k-RAS mutations in endometrial carcinomas with microsatellite instability. J Pathol 2001;193:193–199.
Machin P, Catasus L, Pons C, et al. CTNNB1 mutations and beta-catenin expression in endometrial carcinomas. Hum Pathol 2002;33:206–212.
Palacios J, Catasús L, Moreno-Bueno G, et al., Beta and gamma-catenin expression in endometrial carcinoma. Relationship with clinicopathological features and microsatellite instability. Virchows Arch 2001;438:464–469.
Moreno-Bueno G, Hardisson D, Prat J, et al., Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene 2002;21:7981–7990.
Pollock PM, Gartside MG, Dejeza LC, et al., Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007;26:7158–7162.
Dutt A, Salvesen HB, Chen TH, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA 2008;105:8713–8717.
Byron SA, Gartside MG, Wellens CL, et al., Inhibition of activated fibroblast growth factor receptor 2 in endometrial cancer cells induces cell death despite PTEN abrogation. Cancer Res 2008;68:6902–6907.
Katoh M . Cancer genomics and genetics of FGFR2 (Review). Int J Oncol 2008;33:233–237.
Velasco A, Bussaglia E, Pallares J, et al., PIK3CA gene mutations in endometrial carcinoma. Hum Pathol 2006;37:1465–1472.
Pallarés J, Velasco A, Eritja N, et al., Promoter hypermethylation and reduced expression of RASSF1A are frequent molecular alterations of endometrial carcinoma. Mod Pathol 2008;21:691–699.
Velasco A, Pallares J, Santacana M, et al., Promoter hypermethylation and expression of sprouty 2 in endometrial carcinoma. Hum Pathol 2011;42:185–193.
Pallares J, Martínez-Guitarte JL, Dolcet X, et al., Abnormalities in the NF-kappaB family and related proteins in endometrial carcinoma. J Pathol 2004;204:569–577.
Dolcet X, Llobet D, Pallares J, et al., FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Lab Invest 2005;85:885–894.
Pallares J, Santacana M, Puente S, et al., A review of the applications of tissue microarray technology in understanding the molecular features of endometrial carcinoma. Anal Quant Cytol Histol 2009;31:217–226.
Lo TL, Fong CW, Yusoff P, et al., Sprouty and cancer: the first terms report. Cancer Lett 2006;242:141–150.
Kim HJ, Bar-Sagi D . Modulation of signaling by sprouty: a developing story. Mol Cell Biol 2004;5:441–450.
Turner N, Grose R . Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116–129.
Savagner P, Vallés AM, Jouanneau J, et al., Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol Biol Cell 1994;5:851–862.
Adnane J, Gaudray P, Dionne CA, et al., BEK and FLG, two receptors to members of the FGF family, are amplified in subsets of human breast cancers. Oncogene 1991;6:659–663.
Jang JH, Shin KH, Park JG . Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res 2001;61:3541–3543.
Davies H, Hunter C, Smith R, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res 2005;65:7591–7595.
Hunter DJ, Kraft P, Jacobs KB, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet 2007;39:870–874.
Passos-Bueno MR, Wilcox WR, Jabs EW, et al. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat 1999;14:115–125.
Meulmeester E, Ten Dijke P . The dynamic roles of TGF-β in cancer. J Pathol 2011;223:205–218.
Shirakihara T, Horiguchi K, Miyazawa K, et al. TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J 2011;30:783–795.
Wendt MK, Smith JA, Schiemann WP . Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010;29:6485–6498 .
Schmahl J, Kim Y, Colvin JS, et al. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 2004;131:3627–3636.
Moreno-Bueno G, Rodríguez-Perales S, Sánchez-Estévez C, et al. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene 2003;22:6115–6118.
Acknowledgements
This study was supported by grants FIS PI070276, FIS PI100922, 2009SGR794, RD06/0020/1034 and programa de intensificación de la investigación, Instituto Carlos III. XD holds a postdoctoral fellowship from Fondo de Investigaciones Sanitarias, Ministerio de Sanidad y Consumo (CP05/00028). Tumor samples were obtained with the support of Xarxa catalana de Bancs de Tumors, the Tumor Banc Platform of RTICC and RD09/0076/00059.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Gatius, S., Velasco, A., Azueta, A. et al. FGFR2 alterations in endometrial carcinoma. Mod Pathol 24, 1500–1510 (2011). https://doi.org/10.1038/modpathol.2011.110
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.110
Keywords
This article is cited by
-
Nuclear Fibroblast Growth Factor Receptor Signaling in Skeletal Development and Disease
Current Osteoporosis Reports (2019)
-
Safety, pharmacokinetic, and pharmacodynamics of erdafitinib, a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients with advanced or refractory solid tumors
Investigational New Drugs (2018)
-
Clinical significance of fibroblast growth factor receptor 2 expression in patients with residual rectal cancer after preoperative chemoradiotherapy: relationship with KRAS or BRAF mutations and MSI status
Tumor Biology (2016)
-
Endometrial cancer: redefining the molecular-targeted approach
Cancer Chemotherapy and Pharmacology (2015)
