
Abstract
Myxoid/round cell liposarcoma is characterized by the recurrent translocations t(12;16)(q13;p11) and, less commonly, t(12;22)(q13;q12), which fuse FUS or EWSR1, respectively, to DDIT3 on chromosome 12. Although a number of different variant breakpoints have been described, greater than 90% of all cases have one of the three different FUS–DDIT3 fusions, which may have clinical significance. To identify the individual breakpoints, a sequence-specific assay such as reverse transcription-PCR (RT-PCR) is needed. In this study, we optimized primer design to develop an RT-PCR assay for the detection of the most common translocations in formalin-fixed paraffin-embedded tissue specimens. We compared our assay with primers previously published for testing formalin-fixed paraffin-embedded specimens and achieved the most consistent results with our primers. We obtained RNA from 32 MLS cases, of which 27 carried one of the three common FUS–DDIT3 chimeric transcript types. Four of the negative cases were from very small biopsies with very low RNA concentration. One case was consistently negative by RT-PCR, but showed a FUS rearrangement by fluorescent in situ hybridization, suggesting that it may harbor one of the rarer FUS–DDIT3 chimeric types. In addition to the common fusions, our assay also identified a novel FUS–DDIT3 fusion between exon 9 of FUS and exon 3 of DDIT3 in one of the cases.
Similar content being viewed by others


Main
Myxoid/round cell liposarcoma represents ∼10% of all adult sarcomas and ∼33% of all liposarcomas.1 It tends to affect the extremities of young adults, and is usually treated by surgical resection, beyond which few therapies are available. A higher grade, defined partially by an increasing percentage of round cell component, is associated with a worse prognosis.2, 3 Conventional myxoid liposarcomas have an 80–90% 12-year survival, whereas tumors with a significant round cell component have an ∼50% 12-year survival.4 Conventional myxoid liposarcoma classic histology consists of primitive nonlipogenic mesenchymal cells, chicken wire vasculature, and a myxoid background, but the morphological spectrum of tumors ranges from bland and hyalinized to a more chondroid stroma, and includes the higher-grade cellular round cell liposarcoma.1 Lipoblasts may be seen, but are not required for diagnosis. The differential diagnosis includes other myxoid tumors including myxofibrosarcoma and myxoid chondrosarcoma, as well as lipoblastoma. The hypercellular round cell variants may be confused with other cellular small round cell tumors, especially in smaller biopsies.
Myxoid/round cell liposarcoma is characterized by a recurrent and virtually diagnostic translocation and gene fusion, the t(12;16)(q13;p11), seen in over 90% of cases, which fuses the 5′ half of the FUS gene on chromosome 16 with the entire reading frame of the DDIT3 gene on chromosome 12.2, 5, 6 A much smaller fraction of myxoid liposarcoma cases harbor a similar variant translocation and gene fusion, the t(12;22)(q13;q12), which fuses the EWSR1 gene to the DDIT3 gene.7 FUS and EWSR1 are similar genes, ubiquitously expressed, with a transcriptional activation domain in their 5′ end that is fused to the entire coding region of DDIT3, which encodes an apparent DNA-binding and dimerization domain. This novel chimeric transcription factor is oncogenic for myxoid liposarcoma and inhibits adipocytic differentiation.8, 9, 10 Translocations variably include portions of the RNA-binding domain of FUS or EWSR1, and the oncogenic gene fusion may alter RNA splicing.11
To date, at least 10 different mRNA breakpoints have been described for FUS–DDIT3 (Figure 1) and at least 4 for EWSR1–DDIT3.2, 12, 13 Most cases of myxoid liposarcoma are one of three different FUS–DDIT3 chimeric types, including varying portions of FUS. The other variants are rare and most have only been reported once or twice. Two studies have looked at the clinical impact of the FUS–DDIT3 fusion variants with conflicting results, and no strong clinical significance has been seen in either study.2, 12 However, recent data using a novel chemotherapeutic agent, trabectedin, suggest a possible role for gene fusion variant in predicting a response to therapy.

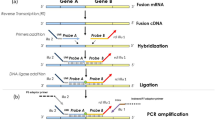
Primer location and expected PCR product size. The top half of this figure shows the location of our FUSEx5-F primers and expected amplicon size from the type 2, 4, and 10 FUS-DDIT3 fusions. The middle panel shows where our FUSEx7-F primers bind and the expected amplicon size from types 1, 3, 6, and 7. Note that the types 3 and 6 and 1 and 7 give the same product. The bottom panel shows the previously reported FUS–DDIT3 fusions not expected to amplify with our primers. Types 5 and 9 do not have a binding site for one of our forward primers. Type 8 fusion could theoretically amplify with our primers, but a 726 bp product is unlikely to be amplified from FFPE tissue.
Trabectedin (ET-743) is a compound isolated from a sea squirt, which binds to the minor groove of DNA, and may act by modifying gene expression or nucleotide excision repair.14 Many patients with myxoid liposarcoma have responded to this drug, and it was noted that patients treated with tumors carrying a type 3 FUS–DDIT3 chimeric transcript showed no response to this agent.15 In addition, an in vitro study of the drug on myxoid liposarcoma cell lines showed that treatment with trabectedin lead to dissociation of the FUS–DDIT3 protein product from chromatin and promotes gene expression consistent with terminal adipogenesis, but this response was only seen in the cell lines with the type 1 FUS–DDIT3 fusion and not in a cell line with the rarer type 8 gene fusion.16 Of note, both the type 3 and type 8 gene fusions have more of the RNA-binding domain from FUS included in the gene fusion. Although these data are incomplete, it suggests that as new therapies are developed, the specific gene fusion type may become relevant, which highlights the need for assays that can differentiate the gene fusion type in the clinical setting.
Formalin-fixed paraffin-embedded tissue is the most commonly available diagnostic specimen in pathology. Routine cytogenetic analysis requires fresh tissue and would not subtype the fusion type. Fluorescent in situ hybridization (FISH) probes are commercially available for DDIT3, FUS, and EWSR1 genes, and can be performed on formalin-fixed paraffin-embedded tissue, but provide no information about the specific fusion partner or mRNA breakpoints. Reverse transcription-PCR (RT-PCR) can be carried out on formalin-fixed paraffin-embedded tissues, but because of RNA degradation, amplicon sizes are small and controls for RNA integrity are needed to avoid false-negative results. Two studies have carried out RT-PCR on formalin-fixed paraffin-embedded tissue in myxoid liposarcoma.12, 17 In this study, we sought to develop a clinically robust RT-PCR assay that could be used on formalin-fixed paraffin-embedded samples for the most common translocations seen in myxoid liposarcoma. We evaluated the primers in the literature and used computational tools to analyze the previously published primers and to design our own, more efficient, PCR primers and assay conditions, which were needed in developing a clinical test.
Materials and methods
Cases
A total of 34 unique histologically confirmed myxoid liposarcoma cases were obtained from the archives at MD Anderson Cancer Center (Table 1) with IRB approval. One case, a known cytogenetically positive t(12;16) case from Texas Children's Hospital was used as a positive control. Most cases showed classic histology. One case was primarily a round cell liposarcoma, and three had a mixed classic and round cell histology. In all, 8 samples were from needle biopsies and 26 samples were from larger resections. All histology samples were reviewed (AJL and WLW) and confirmed as myxoid/round cell liposarcoma. One formalin-fixed paraffin-embedded block was obtained from each case and a 40-μm thick scroll was cut from it; this was followed by H and E staining.
Molecular Methods
RNA was extracted from formalin-fixed paraffin-embedded tissue scrolls using the Ambion RecoverAll kit, as per the manufacturer's instructions (Ambion, Applied Biosystems, Austin, TX, USA). RT was carried out using the Invitrogen Superscript III reverse transcriptase and random hexamers as primers (Invitrogen, Carlsbad, CA, USA). Each RT reaction had 2 μl RNA, 10.1 μl DEPC water, 2.2 μl 5 × RT buffer, 2.2 μl of 10 mM dNTPs, 2.2 μl of 0.1 M DTT, 1.1 μl of random hexamers (50 ng/ml), 0.5 μl of RNase out (40 U/ml), and 0.5 μl of reverse transcriptase. RT was performed at 37°C for 1 h and 95°C for 5 min to inactivate the reaction. cDNA (5 μl) was used in subsequent PCR reactions. Each PCR reaction included 13.875 μl DEPC water, 2.0 μl of PCR buffer, 1.75 μl of dNTPs (10 mM), 2.5 μl of DTT (0.1 M), 0.5 μl of each primer (10 μM working stock each), and 0.125 μl of AmpliTaqGold (Life Technologies, Carlsbad, CA, USA). The following PCR conditions were used an initial denaturation at 94°C for 10 min, 40 cycles at 94°C (30 s), 55°C (60 s), 72°C (2 min), and one final extension at 72°C for 7 min. PCR conditions and the primers used were the same as described in previously published papers and the reagents used are those described in this paper.12, 17 The Invitrogen One-Step RT-PCR kit was used for one-step RT-PCR, as instructed. Amplification of β-actin (234 bp) was used as an RNA integrity control.18 PCR products were visualized on a 2% agarose gel using ethidium bromide staining and sequenced with forward and reverse primers in the ABI PRISM 3100-Avant Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). Electropherograms were interpreted using Chromas Software (Technelysium, Tewantin, QLD, Australia) and the different breakpoints were analyzed using Specialized BLAST Multiple Alignment tool (NCBI).
Primer Design
Three different bioinformatic tools were used to design optimized primers for the detection of the most common FUS–DDIT3 translocations. Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=NcbiHomeAd) was used with the FUS–DDIT3 fusion exon5 to exon 2 and exon 7 to exon 2 cDNA sequences to select 20 candidate primer pairs with an amplicon size of less than 200 bp including all breakpoints, and an annealing temperature of ∼55°C.19 These primer pairs were then submitted to BLAST, and any primer matching greater than 75% of its length to anywhere else in the genome was discarded.20 Lastly, the remaining primer pairs were analyzed for any dimer formation using PerlPrimer.21 Using 57°C as the annealing temperature, only primers with dimerization energy less than 1 kcal/mole were retained. Those with an amplicon size closest to 150 bp were chosen as the final primer pairs (Figure 1). Because of amplicon size and primer location, our primers that were specific for the type 1 and 3 fusions would also give an identical product with the type 7 and 6 fusions, respectively (Figure 1). Primers published in previous papers on myxoid liposarcoma studied in formalin-fixed paraffin-embedded tissues were also analyzed in with the same tools.12, 17 Our final primers were FUSEx5-F: 5′-GCAGAACCAGTACAACAGCA-3′, FUSEx7-F: 5′-GGTGGCTTCAATAAATTTGG-3′, DDIT3Ex3-R: 5′-GGAGAAAGGCAATGACTCAG-3′.
FISH
Sections (5 μm) of available cases were tested for rearrangements using DDIT3 and FUS break-apart FISH probes and the LSI DDIT3–CHOP and LSI FUS–TLS commercially available break-apart probes and the recommended protocols (Vysis/Abbott Molecular, Downers Grove, IL, USA). Briefly, slides were baked at 56°C overnight. The slides were deparaffinized with Hemo-De for 10 min, twice, and with 100% ethanol for 1 min, twice. Protease digestions were carried out by immersing the slide in the pretreatment solution for 10 min at 80°C, washing with purified water at room temperature for 3 min, in protease solution for 20 min at 37°C, and in purified water for 3 min. A volume of 10 μl of the probe mixture (1 μl probe, 7 μl hybridization buffer, and 2 μl water) was hybridized to an ∼1 cm2 area of the slide. Hybridization was carried out in a HyBrite chamber at 80 °C for 5 min and 37°C overnight. Slides were washed four times in 2 × SSC/0.3% NP-40 and visualized. Slides were analyzed using a fluorescence microscope (Olympus BX5, Applied Imaging, San Jose, CA, USA) and Cytovision Genus 3.7 software (Applied Imaging). A minimum of 100 cells was scored for the presence of rearranged signals in a blinded manner (Figure 2).

Case no. 3. The top panel shows the histology of case 3, consistent with a classic myxoid liposarcoma. This case was repeatedly negative by RT-PCR with ours and the published primers. The bottom left panel shows the break-apart FISH for DDIT3, and the bottom right the break-apart FISH for FUS in this case, indicating that this case most likely has a FUS–DDIT3 translocation.
Results
Of the 34 cases extracted (Table 1), β-actin amplicons were obtained in 31, indicating adequate RNA quality. Of these 31 cases, 26 amplified with our PCR primers as expected. The positive control was consistently positive for type 2 fusion. One known EWSR1–DDIT3-positive case was negative, as expected, confirming the specificity of our primers. Of our 25 amplified cases, 13 had type 2 fusion, 6 type 1 (or 7) fusion, and 5 type 3 (or 6) fusion. We also identified one novel variant fusion (discussed below). DDIT3 rearrangements were confirmed by FISH in all positive cases (Table 1).
Of the five cases that did not undergo amplification using our primer pairs, four were from very small biopsies that yielded a very small amount of RNA. An alternative explanation for the negative amplification results is that these samples may carry a variant FUS–DDIT3 or and EWSR1–DDIT3 gene fusion that would not be detected with our primers. Because of the small amount of tissue, we were unable to test either hypothesis using FISH. One case, MLS-3 had an adequate amount of RNA and amplified β-actin, but failed to amplify with our primers. Histology was consistent with myxoid liposarcoma (Figure 2). Break-apart FISH for DDIT3 and FUS indicated that both genes were rearranged, suggesting a FUS–DDIT3 rearrangement (Figure 2). It may be possible that this case has one of the rare variant translocations or a novel and not previously described FUS–DDIT3 fusion that would not amplify with our primers. However, this was the oldest case of our series that showed β-actin amplification (Table 1), and perhaps, not enough FUS–DDIT3 mRNA was sufficiently preserved.
Primers previously published were also examined, but were found to be less sensitive. The nested primers from Hisaoka et al17 were designed toward exons 5 and 2 of the FUS–DDIT3 fusion transcripts and would, therefore, produce an amplicon larger than 200 bp from the type 1 and 3 chimeric transcripts (Figure 1 and Table 2). A total of 11 cases were studied using these primers and with the conditions published in their paper, and only three amplified giving a ladder-type product. Only type 2 fusions were amplified, and not all type 2 fusions tested were positive using this assay (Figure 3 and Table 1). No type 1 or 3 fusions were amplified by these primers. In addition, our primer pairs gave a stronger PCR product, indicating a more robust amplification (Figure 3). When Hisaoka et al's primers were analyzed using Primer-BLAST, NCBI-BLAST, and PerlPrimer, both of their primer pairs, the internal and the external, showed significant nonextensible dimer formation at greater than 1 kcal/mole (Table 2). This type of dimer formation would not be expected to consume excess primer, but may decrease PCR efficiency.

Representative PCR results. The upper left of this figure shows the amplification using the nested primers from Hisaoka et al. Only three of the four type 2 fusions amplified and gave a confusing ladder-like amplification. Case 11, which is strongly amplified by our primers (middle left panel), is not amplified by these primers, and none of the cases with type 1 or 3 fusions were amplified by the nested primers (cases 4,8,17,18, and 19), but were robustly amplified by our primers (bottom left panel for cases 17,18, and 19 shown here). The upper right panel is representative of the results we obtained from the primers published in Bode-Lesniewska et al. Only the type 2 fusions amplified and gave very weak bands on the gel. These same cases (cases 36 and +C) gave strong results with our primers (bottom right panel). In addition, none of the type 1 or 3 fusions were amplified with the Bode-Lesniewska primers (case 34 shown here), whereas the same cases were strongly amplified with our primers (bottom right). The right half of the bottom right panel also shows the size difference between a type 1 fusion (129 bp; case 29) and a type 3 fusion (162 bp; case 34).
The primers from Bode-Lesniewska et al12 target both exons 7 and 5 of FUS and have products that would be expected to amplify genes from formalin-fixed paraffin-embedded tissue (Table 2). However, we had very weak signals from the cases that were type 2 fusions, and no amplification was specified from the type 1 or 3 fusions carried out using the Invitrogen one-step RT-PCR kit and cycling conditions in their paper (Figure 3). Primer-BLAST, BLAST, and PerlPrimer analysis of the Bode-Lesniewska et al primers showed that the primers of exons 5 and 7 in FUS have significant overlap with other transcripts, which may reduce their efficiency, and the FUS exon 7 primer and the reverse primer have significant nonextensible dimer formation, which could also be a possible cause of decreased PCR efficiency (Table 2).
In the course of our investigations, one case, MLS-9, repeatedly showed an unexpected size product of about 210 bp (Figure 4). None of the previously published translocations between FUS and DDIT3 would be expected to generate this amplicon size. This case had characteristic myxoid liposarcoma histology (Figure 4). The cDNA product was sequenced and found to be a new FUS–DDIT3 fusion involving the end of exon 9 of FUS to the fifteenth nucleotide of the third exon of DDIT3 (Figure 4). The frame was maintained from the end of exon 9 to the start ATG codon of DDIT3, consistent with the previously reported FUS–DDIT3 chimeric transcripts. If the 3′ end of exon 9 in FUS were fused to the 5′ end of the third exon of DDIT3, the frame would not be retained. Therefore, the first 14 bp of DDIT3 must be removed to produce a functional fusion protein. Whether this results because of a DNA breakpoint right at or near these nucleotides or because of alternative RNA splicing is unknown. The two immediate nucleotides upstream of the DDIT3 breakpoint are AG (the canonical 3′ splice site), suggesting that this chimeric mRNA results from alternative splicing of the primary fusion transcript. On the basis of the previously published FUS–DDIT3 fusion transcripts, we propose that this be called the type 11 fusion transcript (Figure 5).

Case no. 9. The upper left panel shows the classic myxoid liposarcoma histology in this case. The upper right panel shows the PCR result for this case (far right lane), at an unexpected size of ∼210 bp. Case 8, 162 bp, a type 3 fusion, is immediately to the left of case 9. The PCR product from case 9 was sequenced (bottom panel), and the end of exon 9 (last 5 codons shown in blue text), was continuous with nucleotide 15 of exon 3 of DDIT3 (black text) and in-frame with the ATG start codon of DDIT3 (red text), as seen in all other reported FUS–DDIT3 fusions.

Classification of FUS–DDIT3 fusions. Shown on the top of this figure are the exon structures of the wild-type FUS and DDIT3 genes. The ATG start codon of DDIT3 is shown with a hatch above exon 3. The exon structure of each of the different FUS–DDIT3 fusions is shown below. For the type 1–4 fusions, they are sometimes reported in the literature by the exons fusion (that is, 7–2, 5–3). Our new fusion is proposed as type 11, which fuses the 3′ end of exon 9 to a sequence internal to exon 3 of DDIT3, but still upstream of DDIT3's start codon.
Discussion
In this study, we developed a set of primers that can reliably detect the most common FUS–DDIT3 translocations seen in myxoid/round cell liposarcoma, and discovered a new FUS–DDIT3 variant translocation. Our assay readily distinguishes the three most common breakpoints in FUS–DDIT3, types 1, 2, and 3 translocations. In our study, as the relevant literature would indicate, we most often identified the type 2 translocation, which fuses the fifth exon of FUS to the second exon of DDIT3.2, 12 However, although our assay differentiates whether exon 8 or 7 of FUS is fused to exon 2 or 3 of DDIT3, these primers would not distinguish the splice variants that are missing a portion of exons 5 and 6, (fusion types 6 and 7) (Figure 5), given the limitations of RT-PCR from paraffin-embedded tissue testing. Therefore, the exact variants reported by us and others may be limited by primer choice. To detect these transcripts, primers would be required upstream of exon 5, which would amplify a product larger than that is routinely amplifiable from formalin-fixed paraffin-embedded tissue. In addition, no known significance has been attached to these variant transcripts. In five publications totaling 149 positive cases, 97 (65%) were mentioned to be the exon 5–exon 2 fusions (presumptive type 2), 33 (22%) as exon 7–exon 2 fusions (presumptive type 1), and 13 (9%) exon 8–exon 2 fusions (presumptive type 3).2, 12, 17, 22, 23 The other cases were either a rare novel variant or one of four (3%) cases of a EWSR1–DDIT3 fusion. However, another study using frozen tissue and longer-range PCR from the first four exons of FUS revealed that a number of the exon 7 and exon 8 translocations to DDIT3 often had a portion of exons 5 and 6 spliced out (the type 6 and 7 fusions).24 Also seen in this paper was the fact that many of the tumors had more than one transcript present, with type 6 and type 2 seen simultaneously in a number of cases and one case with type 1, type 6, and type 7 transcripts. Therefore, it is possible that cases determined to be type 1 or type 3 using either our primers or those previously published may also have splice variants that are either type 7 or 6 fusion, respectively, and both splice variants may be seen in the same tumor. Regardless, our assay can reliably determine whether exon 5 of FUS is fused to DDIT3 (the most common type 2 fusion), or whether exons 7 or 8 of FUS are fused to DDIT3.
In this study, we designed a set of primers using different computational tools to increase our primer efficiency. If an RT-PCR assay, which determines FUS–DDIT3 fusion type, is going to be used clinically on formalin-fixed paraffin-embedded tissue, the most efficient and consistent amplification would be needed, as RNA integrity is impaired by formalin fixation and processing. It is often difficult to amplify RNA over 200 bp obtained from formalin-fixed paraffin-embedded tissue, and we include a 234 bp β-actin mRNA control for RNA integrity. We also compared our primers with two previously published primer sets used on formalin-fixed paraffin-embedded tissue.12, 17 Although the previously published primers often worked in most cases, amplification was both more robust and consistent using our newly designed primers. The nested primers from Hisaoka et al17, also would amplify a number of fusions at greater than 200 bp and may miss some of the splice variants or variant fusions in samples with more degraded RNA. We also found some predicted dimerization that may reduce their efficiency. The primers from Bode-Lesniewska et al12 would be expected to amplify the most common fusions from formalin-fixed paraffin-embedded tissue; however, they failed to amplify a number of the cases used in this study. Our analyses suggested a number of variables that may have reduced their efficiency. However, we did not perform the assay with precisely the same reagents indicated in their paper. The subtle change of reagents may have made a difference in efficiency as well. Given the computational tools we used to reduce dimer formation and to reduce off target effects, we expect that our primers would work under varying conditions, and we have shown here that they yield highly reproducible results.
In the course of our studies, we also identified a new FUS–DDIT3 variant, reported never before, between exon 9 of FUS and exon 3 of DDIT3. We are proposing this be called as the type 11 variant of FUS–DDIT3 (Figure 5). This is based on the nine variants published by Panagopoulous et al.13 Since that publication, another variant has been reported between exon 6 of FUS and exon 2 of DDIT3, which we are proposing should be designated as type 10 variant.2, 22 Even though other variants have only been reported in rare cases in the literature, the new variant described here and elsewhere highlight the different possible FUS and DDIT3 fusions seen in myxoid liposarcoma. Whether any of these variants have any clinical significance is not known at this time, highlighting the importance of identifying and reporting cases carrying rare variants. Our variant case showed no unusual histology, and previous publications have not identified any histological differences for any of the variant translocations, common or rare.22
Although still uncertain, the translocation breakpoint variants may have clinical significance. One variant may be more aggressive than the others, or may respond better to chemotherapy such as trabectedin.2, 12, 15, 16 As newer chemotherapeutic and other treatment strategies are developed for myxoid liposarcoma, confirmation of both the diagnosis and subtyping of the translocation may become part of routine clinical practice and of future research studies to identify the best treatment for each patient in this era of personalized medicine. In conclusion, we have developed a clinically robust RT-PCR assay that can identify and determine all known breakpoints in the most common subtypes of myxoid liposarcoma, and have identified a new FUS–DDIT3 mRNA breakpoint, which needs to be considered in future myxoid liposarcoma studies.
References
Fletcher CDM, Unni KK, Mertens F, (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. IARC Press: Lyon, 2002, 427 pp.
Antonescu CR, Tschernyavsky SJ, Decuseara R, et al. Prognostic impact of P53 status, TLS-CHOP fusion transcript structure, and histological grade in myxoid liposarcoma: a molecular and clinicopathologic study of 82 cases. Clin Cancer Res 2001;7:3977–3987.
Smith TA, Easley KA, Goldblum JR . Myxoid/round cell liposarcoma of the extremities. A clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol 1996;20:171–180.
Dalal KM, Antonescu CR, Singer S . Diagnosis and management of lipomatous tumors. J Surg Oncol 2008;97:298–313.
Rabbitts TH, Forster A, Larson R, et al. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 1993;4:175–180.
Crozat A, Aman P, Mandahl N, et al. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993;363:640–644.
Panagopoulos I, Hoglund M, Mertens F, et al. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996;12:489–494.
Riggi N, Cironi L, Provero P, et al. Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res 2006;66:7016–7023.
Engstrom K, Willen H, Kabjorn-Gustafsson C, et al. The myxoid/round cell liposarcoma fusion oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma phenotype in transfected human fibrosarcoma cells. Am J Pathol 2006;168:1642–1653.
Adelmant G, Gilbert JD, Freytag SO . Human translocation liposarcoma-CCAAT/enhancer binding protein (C/EBP) homologous protein (TLS-CHOP) oncoprotein prevents adipocyte differentiation by directly interfering with C/EBPbeta function. J Biol Chem 1998;273:15574–15581.
Rapp TB, Yang L, Conrad III EU, et al. RNA splicing mediated by YB-1 is inhibited by TLS/CHOP in human myxoid liposarcoma cells. J Orthop Res 2002;20:723–729.
Bode-Lesniewska B, Frigerio S, Exner U, et al. Relevance of translocation type in myxoid liposarcoma and identification of a novel EWSR1-DDIT3 fusion. Genes Chromosomes Cancer 2007;46:961–971.
Panagopoulos I, Mertens F, Isaksson M, et al. A novel FUS/CHOP chimera in myxoid liposarcoma. Biochem Biophys Res Commun 2000;279:838–845.
von Mehren M . Trabectedin—a targeted chemotherapy? Lancet Oncol 2007;8:565–567.
Grosso F, Jones RL, Demetri GD, et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 2007;8:595–602.
Forni C, Minuzzo M, Virdis E, et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol Cancer Ther 2009;8:449–457.
Hisaoka M, Tsuji S, Morimitsu Y, et al. Detection of TLS/FUS-CHOP fusion transcripts in myxoid and round cell liposarcomas by nested reverse transcription-polymerase chain reaction using archival paraffin-embedded tissues. Diagn Mol Pathol 1998;7:96–101.
Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol 2008;39:184–193.
Rozen S, Skaletsky H . Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000;132:365–386.
Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215:403–410.
Marshall OJ . PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004;20:2471–2472.
Huang HY, Antonescu CR . Molecular variability of TLS-CHOP structure shows no significant impact on the level of adipogenesis: a comparative ultrastructural and RT-PCR analysis of 14 cases of myxoid/round cell liposarcomas. Ultrastruct Pathol 2003;27:217–226.
Kuroda M, Ishida T, Horiuchi H, et al. Chimeric TLS/FUS-CHOP gene expression and the heterogeneity of its junction in human myxoid and round cell liposarcoma. Am J Pathol 1995;147:1221–1227.
Willeke F, Ridder R, Mechtersheimer G, et al. Analysis of FUS-CHOP fusion transcripts in different types of soft tissue liposarcoma and their diagnostic implications. Clin Cancer Res 1998;4:1779–1784.
Acknowledgements
We thank Karen Prince for assistance with the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Portions of this study were presented in a platform session at the 2009 USCAP meeting in Boston, MA, USA, 7–13 March 2009.
Rights and permissions
About this article
Cite this article
Powers, M., Wang, WL., Hernandez, V. et al. Detection of myxoid liposarcoma-associated FUS–DDIT3 rearrangement variants including a newly identified breakpoint using an optimized RT-PCR assay. Mod Pathol 23, 1307–1315 (2010). https://doi.org/10.1038/modpathol.2010.118
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.118
Keywords
This article is cited by
-
Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas
Modern Pathology (2021)
-
Diagnosis and Treatment of Myxoid Liposarcomas: Histology Matters
Current Treatment Options in Oncology (2018)
-
Establishment and characterization of a new human myxoid liposarcoma cell line (DL-221) with the FUS-DDIT3 translocation
Laboratory Investigation (2016)
-
NY-ESO-1 (CTAG1B) expression in mesenchymal tumors
Modern Pathology (2015)
-
Extensive lipoma-like changes of myxoid liposarcoma: morphologic, immunohistochemical, and molecular cytogenetic analyses
Virchows Archiv (2015)
