
Abstract
Approximately 10–15% of human cancers do not show evidence of telomerase activity, and a subset of these maintain telomere lengths by a recombination-based mechanism termed alternative lengthening of telomeres (ALT). The ALT phenotype, relatively common in certain sarcomas and germ cell tumors, is very rare in carcinomas. In this study we describe evidence for the ALT phenotype in molecular subclasses of breast carcinoma, specifically a subset of cancers with HER-2 overexpression. Tissue microarrays were created from 71 invasive ductal carcinomas of the breast categorized into subclasses, and telomere lengths were directly assessed using fluorescence in situ hybridization with combined promyelocytic leukemia (PML) protein immunofluorescence. The ALT phenotype was identified in 3 of 21 HER-2-positive cases, but in none of the other 50 cases (P=0.023). This is the first direct observation of this mechanism of telomere maintenance in breast carcinoma unrelated to Li–Fraumeni syndrome. The correlation of the ALT phenotype with HER-2 positivity, both of which involve abnormal DNA amplification, suggests a possible common underlying mechanism. This telomere phenotype confers a poor prognosis in some cancers; two of the three cases in our study showed rapid tumor progression, possibly suggesting that it may adversely affect outcome in breast carcinoma as well. As cancers using the ALT pathway are predicted to be resistant to therapies based on telomerase inhibition, these results may have therapeutic consequences.
Similar content being viewed by others


Main
Breast cancer is the most common cancer among women worldwide, and accounts for 18% of all female malignancies.1 Infiltrating duct carcinoma is the most frequent histological type of breast cancer, accounting for approximately 68% of cases.2 Recently, a breast cancer taxonomy based on gene expression profiling with DNA microarrays has led to the characterization of four main breast cancer subgroups, which seem to arise from distinct cell types with unique biologic functions.3 These groups are: luminal A (defined by a significant expression of estrogen receptor (ER) and luminal epithelium-related genes and low histological grade), luminal B (defined by lower levels of ER expression and higher histological grade), HER-2 positive (defined by a low expression of estrogen-related genes and high expression of HER-2-related genes), and basal-like (defined by a low expression of ER and HER-2-related genes and a high expression of myoepithelium-related genes).3, 4 These subgroups predict prognosis and therapeutic response.3, 5 The low-grade luminal A tumors are usually indolent and sensitive to anti-estrogen therapy. Tumors that are both HER-2 positive and ER positive, including luminal B cancers, are less sensitive to hormonal therapy and often carry a worse prognosis. HER-2-positive cancers, which tend to be of high grade, aggressive, and carry a poor prognosis, respond to trastuzumab, a humanized anti-HER-2 antibody. Basal-like cancers are also associated with a worse prognosis,6 both because of the underlying aggressive behavior and lack of targets for hormonal- or antibody-based targeted therapy. Identifying additional molecular markers that distinguish subsets of these classes may yield more detailed prognostic information or carry therapeutic implications.
One such marker is found in the different ways immortalized cells maintain telomere length. Most human cancers (approximately 85%) express telomerase,7 an enzyme that maintains stable chromosomal telomere length and permits unlimited replication. The remaining 10–15% of human cancers do not show evidence of telomerase activity, and a fraction of these maintain telomere lengths by a recombination-based mechanism termed alternative lengthening of telomeres (ALT). The ALT phenotype is commonly identified in adult sarcomas (such as osteosarcomas and liposarcomas),8, 9, 10 astrocytomas and glioblastomas,8, 11 and testicular germ cell tumors, but is extremely rare in carcinomas.12, 13
At the cellular level, ALT is distinguished by the presence of the so-called ALT-associated promyelocytic leukemia (PML) protein nuclear bodies (APBs) that contain large amounts of extra-chromosomal telomeric DNA, PML protein and other proteins involved in telomere binding, DNA replication, and recombination. APBs may be the site of telomere lengthening, or they may hold the protein complexes required for elongation.14 PML nuclear bodies are normally involved in tumor-suppressor functions such as apoptosis and senescence mediated by p53, and Rb-associated repression of transcription.15, 16 In ALT-positive cell lines, bright telomere fluorescence in situ hybridization (FISH) signals in the APBs co-localize with PML protein immunofluorescence staining. There is a marked telomere length heterogeneity in ALT-positive cells, as well as chromosomal instability because of the fusions of subsets of chromosomes with very long or short telomeres.
A recent study showed that the mechanism of ALT telomere maintenance was common in high-frequency microsatellite-unstable gastric carcinomas (33%);17 ALT has also rarely been described in adrenocortical carcinoma.12 However, the ALT mechanism is extremely rare in carcinomas overall, and when identified, is in cell lines that may not reflect in vivo conditions. Although breast carcinomas expressing telomerase activity have been shown to result in poor clinical outcomes,18 the ALT expression in breast cancer has only been reported once in a patient with Li–Fraumeni syndrome.13 In that case, ALT was detected indirectly using a combination of telomerase assays and Southern blotting. The frequency of the ALT phenotype in molecular subclasses of breast carcinoma has not been systematically evaluated. This study is the first to evaluate ALT expression in breast cancer using direct methods (ie, telomere FISH with combined PML immunofluorescence).
Materials and methods
Institutional Review Board Approval
This study was approved by the institutional review board of The Johns Hopkins Medical Institution.
Case Selection and Tissue Microarray Construction
We reviewed cases of invasive ductal carcinoma, resected and processed at our institution, between the years 2001 and 2007. All of these cases were processed uniformly, in that they were sectioned in a fresh state, and fixed overnight in 10% neutral buffered formalin before processing to ensure 24 to 33 h of formalin fixation. Tumors showing processing artifacts (eg, incomplete sections in which fat did not stick to the hematoxylin and eosin-stained slides), of small size (<1 cm), or treated with neo-adjuvant chemotherapy, were excluded from consideration.
Tissue microarrays were constructed as previously described.19 Each array contained 99 tissue cores, each 1.4 mm in diameter. These were arranged as 9 rows, each with 11 columns. Column 6 consisted of various unrelated control tissues, leaving 90 cores on the array for carcinoma samples. For each carcinoma case, five areas were identified on the hematoxylin and eosin slides, punched from the corresponding donor blocks, and placed on the array. Therefore, each array contained 18 different breast tumors. Among the five samples of each case, we attempted to include normal tissue and carcinoma in situ in one sample if possible, leaving four to five cores of infiltrating carcinoma per case.
Immunohistochemistry Methods and Scoring
Immunohistochemistry for ER, progesterone receptor (PR), and HER-2 were previously performed on all cases as part of a routine panel applied to any infiltrating ductal carcinoma at The Johns Hopkins Hospital. The slides were reviewed by two of the authors (APS and PA) to confirm the reported interpretation. Immunohistochemical labeling was performed using standard methods. ER and PR staining was performed on the Benchmark XT autostainer (Ventana Medical Systems Inc., Tucson, AZ, USA) using I-View detection kit. The antibodies, dilutions, and sources were as follows: ER, monoclonal antibody; 1:1 dilution, Ventana, catalog no. 76O-2596; PR, monoclonal antibody, 1:60 dilution, DAKO, catalog no. M3569. Tumors showing weak, moderate, or strong nuclear labeling for ER or PR in >1% of the cells were considered ER positive or PR positive, respectively. HER-2 immunohistochemistry was performed using the DAKO Herceptest kit according to the manufacturer′s standard protocol. The tumors were scored using established criteria as 0 or 1+ (negative), 2+ (equivocal), and 3+ (positive). FISH analysis for HER-2 amplification was performed on all cases with 2+ (equivocal) immunohistochemistry results using the Path Vysion kit (Des-Plaines, IL, USA). To qualify as HER-2 positive for this study, a case had to show either 3+ (strong positive) staining or a HER-2 FISH amplification ratio of >4.0. Cases with equivocal ratios (1.8–2.2), or low-level amplification (ratios 2.2–4.0) were excluded from this study because of their uncertain clinical significance.
Cytokeratin 5/6 (CK 5/6) immunohistochemistry was performed on whole sections of tumors that were negative for ER, PR, and HER-2 to identify basal-like cases, because labeling can be focal. CK 5/6 staining was performed using the Benchmark XT autostainer (prediluted monoclonal antibody, DAKO, catalog no. M7237). Cases were scored on the basis of the percentage of positive cells into 1+(1–25%), 2+(26–50%), 3+(51–75%), and 4+(76–100%) categories. For this study, cases that showed convincing membranous or cytoplasmic labeling in >25% of the neoplastic cells were considered positive. Cases with equivocal or less extensive labeling, which was difficult to distinguish from biotin artifact, were excluded from the study. Immunohistochemistry for epidermal growth factor receptor (EGFR) was performed on cases negative for ER, PR, and HER-2 to identify additional basal-like cases. EGFR labeling was performed using the monoclonal antibody from Zymed, catalog no. 28005, at 1:50 dilution, and the capillary action HRP/DAB detection system (catalog no. 2402, Signet Laboratories Inc., Dedham, MA, USA). Any strong membranous staining for EGFR was considered a positive result. Cases were scored on the basis of the percentage of positive cells into 1+(1–25%), 2+(26–50%), 3+(51–75%), and 4+(76–100%) categories. In general, positive cases showed labeling in 10–50% of the neoplastic cells.
In addition, p53 (Ventana, monoclonal antibody, catalog no. 760-2542) and Ki-67 (Ventana, monoclonal antibody, catalog no. M7240) immunohistochemistry were performed on the Benchmark XT autostainer. For p53 and Ki-67, only nuclear labeling was scored. For p53, labeling of >30% of the nuclei was considered aberrant overexpression, and labeling of less than 30% of the nuclei was considered negative for aberrant overexpression; 30% labeling of nuclei was considered equivocal. The Ki-67 index reflects the mean index of the 4–5 tumor samples per case.
Telomere-Fish (Tel-Fish) and Telomere/Immunostaining-Fish (Teli-Fish)
As previously described,20 deparaffinized slides were hydrated through a graded ethanol series, placed in deionized water, followed by deionized water plus 0.1% Tween-20 detergent for 1 min. They were then placed in citrate buffer (catalog no. H-3300; Vector Laboratories, Burlingame, CA, USA), and steamed for 14 min (Black and Decker Handy Steamer Plus; Black and Decker Corp., Towson, MD, USA), removed, and allowed to cool at room temperature for 5 min. They were then placed in PBS with Tween (PBST) (catalog no. P-3563; Sigma Chemical Co., St Louis, MO, USA) for 5 min. The slides were thoroughly rinsed with deionized water, followed by 95% ethanol for 5 min, and then air-dried. Twenty-five microliters of a Cy3-labeled telomere-specific peptide nucleic acid (PNA) (0.3 μg/ml PNA in 70% formamide, 10 mmol/l Tris, pH 7.5, 0.5% B/M Blocking reagent (catalog no. 1814-320; Boehringer-Mannheim, Indianapolis, IN, USA)) was applied to the sample, which was then coverslipped, and denaturation was performed by incubation for 4 min at 83 °C. The slides were then moved to a dark, closed container for hybridization at room temperature for 2 h. Coverslips were then carefully removed and the slides were washed twice in PNA wash solution (70% formamide, 10 mmol/l Tris, pH 7.5, 0.1% albumin (from 30% albumin solution, catalog no. a-7284; Sigma)), followed by 3 × 5-min washes in TBST.
At this point, slides were either counterstained with 4′-6-diamidino-2-phenylindole (DAPI) (500 ng/ml in deionized water, Sigma Chemical Co., Cat no. D-8417) for 1 min at room temperature, mounted with Prolong anti-fade mounting medium (catalog no. P-7481; Molecular Probes Inc., Eugene, OR, USA), and imaged, or were further processed for indirect immunofluorescence as follows. The slides were rinsed in PBST followed by application of primary antibody (anti-smooth muscle actin, Dako, Carpinteria, CA, USA, Cat no. m0851 or anti-PML antibody, Dako Cat no. PG-M3 diluted 1:100) and incubated overnight at 4 °C. They were then rinsed in PBST followed by application of goat anti-rabbit or goat anti-mouse IgG fraction Alexa Fluor 488 fluorescent secondary antibody (Molecular Probes Cat nos. A-11034 and A-11001, respectively) diluted 1:100 in Dulbecco's PBS, and incubated at room temperature for 30 min. They were then rinsed in PBST, and thoroughly rinsed in deionized water. The slides were drained and stained with DAPI, and were then rinsed well in deionized water, drained, mounted with Prolong anti-fade mounting medium (catalog no. P-7481, Molecular Probes Inc.), coverslipped, and imaged or stored at 4 °C until used. The PNA probe complementary to the mammalian telomere repeat sequence was obtained from Applied Biosystems (Framingham, MA, USA), and has the sequence (N-terminus to C-terminus) CCCTAACCCTAACCCTAA with an N-terminal covalently linked Cy3 fluorescent dye. As a positive control for hybridization efficiency, an FITC-labeled PNA probe having the sequence ATTCGTTGGAAACGGGA with specificity for human centromeric DNA repeats (CENP-B binding sequence) was also included in the hybridization solution.21
Microscopy
The slides were imaged using Nikon 50i epifluorescence microscope equipped with X-Cite series 120 illuminator (EXFO Photonics Solutions Inc., Ontario, CA, USA) and a 100 × /1.4 NA oil immersion Neofluar lens. Fluorescence excitation/emission filters were as follows: Cy3 excitation, 546 nm/10 nm BP; emission, 578 nm LP (Carl Zeiss Inc.); DAPI excitation, 330 nm; emission, 400 nm through an XF02 fluorescence set (Omega Optical, Brattleboro, VT); Alexa Fluor 488 excitation, 475 nm; emission, 535 nm through a combination of 475RDF40 and 535RDF45 filters (Omega Optical). Grayscale images were captured for presentation using Nikon NIS-Elements software and an attached Photometrics CoolsnapEZ digital camera, pseudo-colored and merged. Integration times typically ranged from 500 to 800 ms for Cy3 (telomere) and FITC (centromere) signal capture, 50 to 100 ms for the DAPI counterstain, and 100 to 200 ms for Alexa Fluor 488-conjugated antibodies.
The arrays were assessed for the presence of the ALT phenotype by two authors (APS and AKM). The ALT-positive cases were identified by large, very bright, intra-nuclear foci of telomere FISH signals marking APBs throughout the tumor cells, confirmed by co-localization of PML protein by combined PML immunofluorescence. Areas showing necrosis were excluded from consideration.
Results
Case Characterization
The 71 breast carcinoma cases on the tissue microarrays were categorized into one of the four groups based upon previously validated and accepted immunohistochemical surrogate profiles.22, 23 A table with details of all 71 cases has been published previously.24 In all, 17 cases were immunoreactive for ER and negative for HER-2, and were classified as luminal A tumors. The luminal A cases showed nuclear labeling for ER in at least 70% of the invasive carcinoma cells. Of the 21 HER-2 positive cases identified by 3+ immunohistochemical stain and/or amplification ratio >4.0 by FISH, 14 were negative for ER and PR and were considered classic HER-2 cases; 11 of these cases were reported as 3+ by immunohistochemistry, and the remaining 3 had FISH amplification ratios of 19.6, 4.1, and 14.2. There were 7 of 21 HER-2-positive cases that were immunoreactive for ER and/or PR, though to a lesser degree than the luminal A cases. These cases were therefore considered luminal B tumors. Five of these cases showed 3+ HER-2 staining by immunohistochemistry, and the remaining two had FISH amplification ratios of 5.6 and >4.0 (not specifically quantified).
Among the 33 cases that were negative for ER, PR, and HER-2 (triple-negative carcinomas), 8 showed staining for CK 5/6 and EGFR, 9 stained for CK 5/6 but not EGFR, and 4 stained for EGFR but not CK 5/6. These 21 cases were categorized as basal-like carcinomas on the basis of published criteria. The remaining 12 cases, which were negative for ER, PR, HER-2, CK 5/6, and EGFR, were considered unclassifiable triple-negative carcinomas. Of note, the category of infiltrating ductal carcinoma, considered ‘normal breast-like’ by gene expression profiling, is poorly defined and lacks a validated immunohistochemical surrogate profile, and was not included in this study.
Patient Demographics, Tumor Grade, and Stage
Age, ethnicity, grade, and lymph node status of the carcinoma subclasses are summarized in Table 1, a version of which has been published previously.24 Luminal A cancers occurred in older patients and were more likely to be of lower Elston grade (1 and 2) (ages of luminal A vs luminal B P=0.040, vs HER-2 P=0.055, vs basal-like carcinoma P=0.0035, vs unclassifiable triple-negative carcinoma P=0.021) (grade of HER-2, basal-like carcinoma, and unclassifiable triple negative carcinoma each vs luminal A P<0.001, grade of luminal B vs luminal A P=0.21). In addition, a higher percentage of basal-like carcinomas and unclassifiable triple-negative carcinomas occurred in African-American patients (combined triple-negative carcinomas vs all others P=0.046). Both of these trends are consistent with published literature.22, 25, 26
The p53 and Ki-67 Indices
The results of p53 overexpression and Ki-67 indices are summarized in Table 2, a version of which has been published previously.24 In total, 2 of 17 luminal A cases, 4 of 7 luminal B cases, and 8 of 14 HER-2 cases overexpressed p53, along with 10 of 21 basal-like carcinomas and 8 of 12 unclassifiable triple-negative carcinomas. The p53 expression pattern between each of the HER-2, basal-like carcinoma, and unclassifiable triple-negative carcinoma groups, compared with the luminal A cancers, was significant (P<0.05), but there was no significant difference among the three former groups. Luminal B cancers showed p53 overexpression more frequently than luminal A cancers (P=0.03).
The basal-like carcinoma and unclassifiable triple-negative carcinoma groups had the highest proliferation indices among the five groups. The mean Ki-67 index for the basal-like carcinoma group was 67.5%; the mean for the unclassifiable triple-negative carcinoma group was 61.5%. HER-2-positive cases had intermediate proliferation indices: the mean Ki-67 index for the HER-2 cases was 36.5%, whereas the mean for the luminal B cases was 35.1%. Using analysis of variance, these groups showed a statistically significant difference in mean Ki-67 indices (P<0.001). As expected, luminal A cases, which included more cancers of lower grade, had the lowest Ki-67 indices. The mean Ki-67 index average for the luminal A cases was 8.6% (luminal A vs all others P<0.001).
Assessment for Alternative Lengthening of Telomeres (ALT)
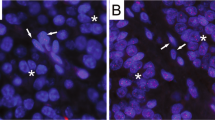
The ALT phenotype was identified in 3 of 21 HER-2-positive cases (luminal B and HER-2), and was further verified by co-localization of unusually bright telomere FISH signals with PML protein, as revealed by simultaneous anti-PML immunofluorescence. A comparison of an ALT-positive case with an ALT-negative case is shown in Figure 1. The arrow shows APBs indicative of the ALT phenotype; in the example of the ALT-negative case, the arrowheads indicate robust telomeres present in non-tumor stromal cells, adjacent to tumor cells showing telomere shortening by dim telomere signal. Figures 2a–c show confirmation of an ALT-positive case, by identification of PML protein co-localization with collections of telomere DNA to form APBs. The inset highlights the typical targetoid appearance of an APB with a peripheral rim of PML protein. For comparison in Figures 2d–f, the PML protein immunofluorescence does not overlap with telomere signals in an ALT-negative case.

(a) An ALT-positive case. The arrow indicates ALT-associated promyelocytic leukemia protein bodies (APBs) in a tumor cell. (b) An ALT-negative case, which shows telomere shortening. For comparison, the arrowheads indicate non-tumor stromal cells that show robust telomeres, as would be expected in normal tissue. The background cellular DNA is stained blue by DAPI, telomere DNA is stained red by telomere probe, and centromeres are stained green by centromere probe. Original magnification × 400.

Co-localization of telomere DNA and promyelocytic leukemia (PML) protein in the ALT phenotype (a–c) . An ALT-positive case; the inset in (c) highlights the typical targetoid appearance of an APB with a peripheral rim of PML protein. (d–f) An ALT-negative case; in such cases, APBs are absent and PML protein does not co-localize with telomere DNA. The background cellular DNA is stained blue by DAPI, telomere DNA is stained red by telomere probe, and PML protein is stained green by anti-PML antibody. Original magnification × 400, inset × 1000.
The details of the three ALT-positive cases are included in Table 3. The HER-2 amplification ratios of the three cases were 5.6, 19.6, and 4.1, respectively. All three tumors were Elston grade 3; two out of three were negative for p53 overexpression by immunohistochemistry. Histologically, they showed pleomorphic nuclei and a high proliferative rate with atypical mitotic figures, but were indistinguishable from ALT-negative HER-2-positive cases based on routine hematoxylin and eosin staining. As is consistent with descriptions of the ALT phenotype in the literature, our three cases showed scattered APBs throughout the tumor cells in all respective tissue cores, but APBs were not observed uniformly in every cell.
None of the 17 luminal A cases, 21 basal-like carcinoma cases, or 12 unclassifiable triple-negative carcinoma cases showed the ALT phenotype. This difference was statistically significant (P=0.023) using Fisher's exact test.
Follow-Up on HER-2-Positive Cases
Of the three ALT-positive cases, Case 1, a 34.6-year-old female with a tumor categorized as luminal B, died of disease after 1.25 years. Case 2, a 45-year-old female with a classic HER-2 tumor, was alive without disease at 3.5 years. Case 3, a 54.7-year-old female with a classic HER-2 tumor, died of disease after 9 months.
Of the six ALT-negative luminal B cases, two died of disease with an average survival duration of 3.6 years, and four were alive without evidence of disease with an average follow-up interval of 4.8 years.
Of the 12 ALT-negative classic HER-2 cases, 2 died of disease with an average survival duration of 1.9 years; 1 was alive with disease at 2.2 years; 8 were alive without evidence of disease with an average follow-up interval of 3.5 years; and 1 patient was alive at 6.7 years but the disease status was unknown.
Discussion
Maintenance of telomeres is an essential component of cell immortalization, and is primarily accomplished through the enzymatic activity of telomerase.27 It has been shown that introducing telomerase function in normal human fibroblasts can extend their in vitro replicative potential indefinitely.28 Furthermore, telomerase is known to be lacking or far less active in normal somatic cells compared with the majority of human tumors studied.14, 29 Novel chemotherapeutics targeting telomerase are being developed to exploit this differential in activity between normal somatic and malignant cells.30 However, tumors using telomerase-independent mechanisms of telomere maintenance, such as ALT, would be expected to be resistant to such strategies;31 such drugs might even provide evolutionary pressure to select for ALT-positive tumor clones.32
To date, the ALT mechanism has been found most prominently in sarcomas.8, 9, 10 It has been suggested that telomerase expression may be more stringently suppressed in mesenchymal tissues,32, 33 potentially explaining the higher frequency of ALT in sarcomas. The ALT-positive tumors can later develop into telomerase-expressing tumors;32 furthermore, both mechanisms of telomere maintenance can also be expressed in the same cell line.9, 10
The ALT phenotype is now known to be a recombination-based mechanism, after telomere tagging experiments showed unique sequences from an integrated plasmid in the telomere of one chromosome increasing in number and appearing at the ends of other chromosomes.34 Furthermore, ALT-positive cells show high frequencies of post-replicative telomeric exchanges.35 Cell lines expressing the ALT phenotype show a wide range of telomere lengths, from a few hundred to over 50 kilobase pairs.14 Because of the marked heterogeneity of telomere length in ALT, there is often chromosome instability due to fusions involving subsets of chromosomes with very short or very long telomeres. The telomeres of ALT-positive cells undergo gradual erosion primarily because of incomplete replication, but there can be rapid lengthening or shortening as well.36 Analysis of the cell cycle has shown that APBs are in highest quantity during G2 phase, which seems to correspond with the elongation of telomere length in ALT-positive tumor cells.37
ERBB2 is a proto-oncogene localized to chromosome 17q that encodes a transmembrane tyrosine kinase growth factor receptor; amplification of the ERBB2 gene resulting in overexpression of the HER-2 receptor has been identified in 15–20% of invasive breast cancers.38 Several other genes implicated in the pathogenesis of breast carcinoma are located on chromosome 17, including the tumor-suppressor genes p53 and BRCA1, and the gene for topoisomerase IIα.39 It is therefore not surprising that up to 93% of breast tumors have whole chromosome 17 copy-number changes.40 Reinholz et al39 concluded in their recent review that chromosome 17 monosomy is more common in non-invasive and preinvasive breast lesions, whereas polysomy 17 is more common in invasive carcinomas. These findings support the notion that the underlying degree of chromosomal instability may correlate with the malignant progression of ductal carcinoma in situ to invasive carcinoma.41 However, HER-2 overexpression in invasive breast cancer typically results from gene amplification independent of polysomy 17, and polysomy 17 alone does not predict a therapeutic response to anti-HER-2 therapy in the absence of increased gene copy number.39 How ERBB2 gene amplification is promoted in HER-2 breast cancers is not well understood at this point.
In this small study, the ALT phenotype, which is virtually never observed in other carcinomas, was observed rarely in breast cancer, but it did occur preferentially in a subset of breast cancers with HER-2 overexpression. It is known that HER-2-positive breast cancers are marked by high-level gene amplifications, suggestive of a greater degree of genomic complexity and alteration.3, 39, 42 Recent studies have assessed allelic imbalance as a marker of genome-wide instability, and have shown numerous differences in tumors with amplification of the HER-2 receptor versus those without.43 Similarly, global copy number changes in several chromosomes, including gains of 1q, 8q, and 20q, and losses of 18q, 13q, and 3p, have been shown in a large proportion of HER-2-amplified breast cancers using comparative genomic hybridization.44 The ALT mechanism seems to operate through break-induced replication and recombination events.14 Such a process could lead to free chromosome ends, which then participate in end-to-end associations and induce breakage-fusion-bridge cycles, resulting in an increased number of complex chromosomal rearrangements,45 which could theoretically contribute to the genome-wide instability that has been observed in HER-2-amplified breast cancers. Other evidences do not seem to directly implicate ALT in increases in homologous recombination events.46, 47 Still, the observation that activation of the ALT phenotype can affect other sequences in the genome48 raises the possibility that the ALT may have a role in the genetic pathogenesis of HER-2 amplification in some cases.
We believe that our study is the first direct observation of ALT in breast carcinoma, and it has been previously described only once in a breast cancer from a patient with Li–Fraumeni syndrome.13 In that case, ALT was detected indirectly using a combination of telomerase assays and Southern blotting. As cancers using the ALT pathway are predicted to be resistant to therapies based on telomerase inhibition,31 these results may have therapeutic consequences. This study was not sufficiently powered to detect a difference in prognosis for ALT-positive cases. However, the presence of the ALT phenotype does predict a poor prognosis in some cancers,10 and its prognostic significance in infiltrating ductal carcinoma of the breast should be examined in the future. The rapid tumor progression in two out of the three cases in this study at least suggests that ALT may adversely affect outcomes in a similar manner. Moreover, whether the ALT phenotype has any role in the response of HER-2-positive breast cancers to trastuzumab is a question for further study.
References
McPherson K, Steel CM, Dixon JM . ABC of breast diseases. Breast cancer-epidemiology, risk factors, and genetics. BMJ 2000;321:624–628.
Berg JW, Hutter RVP . Breast cancer. Cancer 1995;75:257–269.
Sotiriou C, Pusztai L . Gene-expression signatures in breast cancer. N Engl J Med 2009;360:790–800.
Sotiriou C, Neo SY, McShane LM, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci USA 2003;100:10393–10398.
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001;98:10869–10874.
Schneider BP, Winer EP, Foulkes WD, et al. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res 2008;14:8010–8018.
Shay JW, Bacchetti S . A survey of telomerase activity in human cancer. Eur J Cancer 1997;33:787–791.
Henson JD, Hannay JA, McCarthy SW, et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res 2005;11:217–225.
Henson JD, Hannay JA, McCarthy SW, et al. Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin Cancer Res 2005;11:5347–5355.
Costa A, Daidone MG, Daprai L, et al. Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res 2006;66:8918–8924.
Hakin-Smith V, Jellinek DA, Levy D, et al. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003;361:836–838.
Else T, Giordano TJ, Hammer GD . Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J Clin Endocrinol Metab 2008;93:1442–1449.
Bryan TM, Englezou A, Dalla-Pozza L, et al. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 1997;3:1271–1274.
Royle NJ, Foxon J, Jeyapalan JN, et al. Telomere length maintenance—an ALTernative mechanism. Cytogenet Genome Res 2008;122:281–291.
Guo A, Salomoni P, Luo J, et al. The function of PML in p53-dependent apoptosis. Nat Cell Biol 2000;2:730–736.
Khan MM, Nomura T, Kim H, et al. PML-RARalpha alleviates the transcriptional repression mediated by tumor suppressor Rb. J Biol Chem 2001;276:43491–43494.
Omori Y, Nakayama F, Li D, et al. Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer Sci 2009;100:413–418.
Poremba C, Heine B, Diallo R, et al. Telomerase as a prognostic marker in breast cancer: high-throughput tissue microarray analysis of hTERT and hTr. J Pathol 2002;198:181–189.
Wu JM, Fackler MJ, Halushka MK, et al. Heterogeneity of breast cancer metastasis: comparison of therapeutic target expression and promoter methylation between primary tumors and their multifocal metastases. Clin Cancer Res 2008;14:1938–1946.
Meeker AK, Gage WR, Hicks JL, et al. Telomere length assessment in human archival tissues: combined telomere fluorescence in situ hybridization and immunostaining. Am J Pathol 2002;160:1259–1268.
Chen C, Hong YK, Ontiveros SD, Egholm M, Strauss WM . Single base discrimination of CENP-B repeats on mouse and human Chromosomes with PNA-FISH. Mamm Genome 1999;10:13–18.
Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006;295:2492–2502.
Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 2004;10:5367–5374.
Subhawong AP, Subhawong T, Nassar H, et al. Most basal-like breast carcinomas demonstrate the same Rb-/p16+ immunophenotype as the HPV-related poorly differentiated squamous cell carcinomas which they resemble morphologically. Am J Surg Pathol 2009;33:163–175.
Fadare O, Tavassoli FA . The phenotypic spectrum of basal-like breast cancers: a critical appraisal. Adv Anat Pathol 2007;14:358–373.
Fadare O, Yeh I . Basal-like breast cancers. Pathol Case Rev 2007;12:143–153.
Counter CM, Avilion AA, LeFeuvre CE, et al. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 1992;11:1921–1929.
Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998;279:349–352.
Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011–2015.
Perry PJ, Arnold JRP, Jenkins TC . Telomerase inhibitors for the treatment of cancer: the current perspective. Expert Opin Investig Drugs 2001;10:2141–2156.
Neumann AA, Reddel RR . Telomere maintenance and cancer -- look, no telomerase. Nat Rev Cancer 2002;2:879–884.
Henson JD, Neumann AA, Yeager TR, et al., Alternative lengthening of telomeres in mammalian cells. Oncogene 2002;21:598–610.
Serakinci N, Hoare SF, Kassem M, et al., Telomerase promoter reprogramming and interaction with general transcription factors in the human mesenchymal stem cell. Regen Med 2006;1:125–131.
Dunham MA, Neumann AA, Fasching CL, et al., Telomere maintenance by recombination in human cells. Nat Genet 2000;26:447–450.
Londoño-Vallejo JA, Der-Sarkissian H, Cazes L, et al., Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res 2004;64:2324–2327.
Wang RC, Smogorzewska A, de Lange T . Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 2004;119:355–368.
Grobelny JV, Godwin AK, Broccoli D . ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci 2000;113:4577–4585.
Ross JS, Fletcher JA . The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells 1998;16:413–428.
Reinholz MM, Bruzek AK, Visscher DW, et al. Breast cancer and aneusomy 17: implications for carcinogenesis and therapeutic response. Lancet Oncol 2009;10:267–277.
Botti C, Pescatore B, Mottolese M, et al. Incidence of chromosomes 1 and 17 aneusomy in breast cancer and adjacent tissue: an interphase cytogenetic study. J Am Coll Surg 2000;190:530–539.
Visscher D, Jimenez RE, Grayson 3rd M, et al., Histopathologic analysis of chromosome aneuploidy in ductal carcinoma in situ. Hum Pathol 2000;31:201–207.
Chin K, DeVries S, Fridlyand J, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006;10:529–541.
Ellsworth RE, Ellsworth DL, Patney HL, et al. Amplification of HER2 is a marker for global genomic instability. BMC Cancer 2008;8:297.
Isola J, Chu L, DeVries S, et al. Genetic alterations in ERBB2-amplified breast carcinomas. Clin Cancer Res 1999;5:4140–4145.
Scheel C, Schaefer KL, Jauch A, et al. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001;20:3835–3844.
Dunham MA, Neumann AA, Fasching CL, et al., Telomere maintenance by recombination in human cells. Nat Genet 2000;26:447–450.
Bechter OE, Zou Y, Shay JW, et al., Homologous recombination in human telomerase-positive and ALT cells occurs with the same frequency. EMBO Rep 2003;4:1138–1143.
Jeyapalan JN, Varley H, Foxon JL, et al. Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum Mol Genet 2005;14:1785–1794.
Acknowledgements
We thank Dr Ty K Subhawong, MD, for critically reviewing this paper, and Dr Peter B Illei, MD, for assisting with determination of HER-2 FISH amplification ratios.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Subhawong, A., Heaphy, C., Argani, P. et al. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod Pathol 22, 1423–1431 (2009). https://doi.org/10.1038/modpathol.2009.125
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2009.125
Keywords
This article is cited by
-
THOR is a targetable epigenetic biomarker with clinical implications in breast cancer
Clinical Epigenetics (2022)
-
Shorter telomeres in luminal B, HER-2 and triple-negative breast cancer subtypes
Modern Pathology (2011)
-
Alternative lengthening of telomeres: models, mechanisms and implications
Nature Reviews Genetics (2010)
