
Abstract
Aurora-A kinase is a cell-cycle-regulating kinase required for chromosomal segregation. Overexpression of Aurora-A kinase has been shown to correlate with tumor proliferation and chromosomal instability. We investigated Aurora-A kinase expression in peripheral blood and bone marrow of 47 chronic lymphocytic leukemia patients and 20 age-matched hematologically healthy subjects. Western blot analysis showed significantly higher Aurora-A levels in chronic lymphocytic leukemia (42 of 47) compared with lymphocytes of healthy subjects. However, Aurora-A mRNA expression in three chronic lymphocytic leukemia patients was similar to or lower than that of healthy control subjects. In 28 of 42 chronic lymphocytic leukemia patients with elevated Aurora-A kinase expression, one or more chromosomal abnormalities were detected, including trisomy 12 in 9 patients and deletion of the ataxia telangiectasia-mutated gene in 9 patients. Aurora-A was also detected in all (100%) chronic lymphocytic leukemia cases by immunohistochemistry, with a nuclear staining pattern. The larger prolymphocytes and paraimmunoblasts showed stronger Aurora-A kinase expression than did small lymphocytes. In contrast, normal bone marrow reactive lymphocytes were negative for Aurora-A with positive histiocytes and immature myeloid cells. Immunostaining for acetylated histone H3 showed a nuclear pattern in all 38 chronic lymphocytic leukemia cases and double labeling showed coexpression of acetylated histone H3 and Aurora-A. In summary, Aurora-A kinase is overexpressed in chronic lymphocytic leukemia cells. The expression of acetylated histone H3 suggests that Aurora-A kinase may be active (functional). Thus, Aurora-A kinase overexpression in chronic lymphocytic leukemia may be involved in the genesis of chromosomal abnormalities and is a potential target for therapeutic intervention.
Similar content being viewed by others

Main
Chronic lymphocytic leukemia (CLL) is the most common type of adult leukemia in the United States and Western Europe. Traditionally, CLL has been described as a disorder characterized by the accumulation of nondividing small lymphocytes, most of which are in the G0/G1 phase of the cell cycle as a result of defective apoptosis.1 However, CLL also has a proliferative component of prolymphocytes and paraimmunoblasts, predominantly found in the proliferation centers of lymph nodes.2 This proliferative component is thought to be important for disease progression. Messmer et al3 recently used a nonradioactive, stable isotope labeling technique to show that CLL is not a static disease, but rather a dynamic process in which the neoplastic cells proliferate and die at appreciable rates, thus suggesting that a correlation may exist between proliferation and progression. Furthermore, Obermann et al4 immunohistochemically assessed the cell-cycle phase distribution of CLL cells and showed that a large number of the neoplastic cells had proliferative potential.
Aurora kinases are mitotic kinases that are especially important in regulating the G2/M phase of the cell cycle and various mitotic events, including centrosomal duplication, mitotic spindle assembly, chromosome segregation, and cytokinesis at the end of telophase.5, 6 Three major Aurora kinases—A, B, and C—have been described, and each serves at different stages of mitosis.7 Altered expression of Aurora kinase proteins has been implicated in the pathogenesis of various cancers, including those of the breast, pancreas, colon, ovary, and urinary bladder.8 Overexpression of Aurora kinases also induces chromosomal instability.9 Aurora-A is overexpressed in aggressive types of non-Hodgkin's lymphoma, including diffuse large B-cell and Burkitt lymphomas.10
Up to 15% of CLL patients have a progressive or accelerated clinical course and these neoplasms often have chromosomal abnormalities, such as trisomy 12, p53 gene mutation, and deletion of the ataxia telangiectasia-mutated (ATM) gene.1 It is possible that deregulation or overexpression of one or more of the Aurora kinases plays a role in inducing chromosomal instability in CLL patients. This suggestion is in accordance with our previous observation that atypical CLL cells with aberrant nuclear morphology are noted in bone marrow aspirate and peripheral blood smears with disease progression.11 Therefore, we investigated the pattern of Aurora kinase A expression in the peripheral blood and bone marrow of patients with CLL.
Materials and methods
Patient Specimens
With the approval of the institutional review board at The University of Texas MD Anderson Cancer Center, we obtained peripheral blood and bone marrow specimens (aspirate, clot, and biopsy) from 47 patients with CLL. We also obtained peripheral blood specimens from 20 age-matched hematologically healthy subjects. The relevant clinical characteristics are shown in Table 1.
The diagnosis of CLL was established in each case using previously published clinical, morphologic, laboratory, and immunophenotypic criteria.1 These criteria included the presence of a B-cell neoplasm composed predominantly of small, mature-appearing lymphocytes. These B-cells expressed CD5, CD19, CD20, and CD23 and monotypic immunoglobulin light-chain. Cytogenetic analyses were performed using conventional G-banding or fluorescence in situ hybridization (FISH), as described previously.12 Our FISH probe set allowed detection of the following genomic aberrations in CLL: deletion of 11q22.3 at the ATM locus, deletion of 17p13.1 at the p53 locus, deletion of 13q14.3 at the D13S319 locus, deletion of 13q34 at the LAMP1 locus, and trisomy 12.
Selection of B Cells by Magnetic Cell Sorting
Peripheral blood mononuclear cells were isolated from anticoagulated blood specimens by Ficoll density gradient centrifugation followed by B-cell selection according to the manufacturer's specifications (B-cell isolation kit II, Miltenyi Biotec, Auburn, CA, USA). We used a phycoerythrin-conjugated CD19 antibody to confirm greater than 90% B-cell enrichment.
Western Blot Analysis
Peripheral blood CD19+ B-cell lysates from patients with CLL and from healthy subjects were used for western blot analysis. A BioRad Protein Assay Reagent Kit (Bio-Rad, Hercules, CA, USA) was used to determine the protein concentration of the cell lysates. Protein electrophoresis was carried out by 10% SDS–polyacrylamide gel electrophoresis, and proteins were transferred to Hybond-P polyvinylidene difluoride membranes (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). The membranes were probed with rabbit anti-human Aurora-A antibody (Abcam Inc., Cambridge, MA, USA; 1:500 dilution) and then reprobed with anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal antibody (Ambion, TX, USA; 1:8000 dilution) to confirm equal loading of protein. A lysate of the MCF-7 breast cancer cell line was used as a positive control.
Immunohistochemical and Immunocytochemical Staining
Immunohistochemical analysis was performed using formalin-fixed, paraffin-embedded sections (4 μm thick) of bone marrow biopsy specimens from 47 patients with CLL and from four control subjects who underwent bone marrow biopsy for staging purposes and had incidental benign lymphoid aggregates. Tissue sections were incubated for 60 min with antibodies specific for total Aurora-A (Novus Biologicals Inc., Littleton, CO, USA; 1:500 dilution), p53 (DO-7; Dako Cytomation Inc., Carpinteria, CA, USA; 1:100 dilution), and acetylated histone H3 (Cell Signaling Technology, Danvers, MA, USA; 1:200 dilution). Immunohistochemical analysis was carried out by using the two-step EnVision+ System-HRP methodology, as described in the product insert (Dako Cytomation Inc.). Bone marrow biopsy specimens of metastatic prostate carcinoma were used as a positive control.
Immunocytochemical analysis for phosphorylated Aurora-A (Abcam Inc.; 1:200 dilution) and total Aurora-A expression was performed on formalin-fixed bone marrow aspirate smears from 15 patients with CLL by using the alkaline phosphatase labeling method, as described previously.13
Quantitative Reverse Transcriptase-PCR for Aurora-A mRNA Expression
A quantitative reverse transcriptase-PCR (RT-PCR) method was used to determine Aurora-A expression in five untreated patients with CLL, five hematologically healthy controls, and cells from four corresponding mantle cell lymphoma cell lines as positive controls. β-Actin mRNA was used as the internal control. Total RNA was extracted from CD19+ B cells using the Trizol method (Invitrogen, Carlsbad, CA, USA). cDNA was prepared with the Quantitect RT Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Aurora-A cDNA was quantified using TaqMan Universal PCR Mastermix (Applied Biosystems, Foster City, CA, USA) and the Aurora-A TaqMan Gene Expression Assay using the manufacturer's specifications. β-Actin was assayed using TaqMan Universal PCR Mastermix with forward (CCCTGGCACCCAGCAC) and reverse (GCCGATCCACACGGAGTAC) primers at 400 nM each and probe (fam-ATCAAGATCATTGCTCCTCCTGAGCGC-bhq) at 100 nM concentrations. Real-time quantitative RT-PCR was performed in an automatic MX3000P real-time RT-PCR thermal cycler (Stratagene, La Jolla, CA, USA). Relative RNA level was reported via standard delta delta Ct (δδ Ct). cDNA purified from normal CD19+ cells was used as calibrator sample (Allcells, LLC, Emeryville, CA, USA).
Statistical Analysis
Fisher's exact test and Wilcoxon rank-sum test were used to determine the association of Aurora-A with chromosomal aberrations and various clinicopathologic parameters. Using CART analysis, an optimal cutoff value for Aurora-A/GAPDH ratio was selected to classify it as ‘normal’ vs ‘high’. Fisher's exact test was used to assess the categorized Aurora-A/GAPDH ratio and various chromosomal aberrations. P-values less than 0.05 were deemed as statistically significant. All analyses were carried out in Splus.
Results
Western Blot Analysis
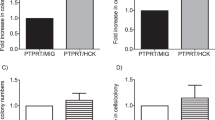
Western blot analysis showed that Aurora-A was expressed in all 47 patients with CLL and in the 20 hematologically healthy subjects. MCF-7 breast cancer cells, reported previously to express high levels of Aurora-A, were used as a positive control and the GAPDH gene as a loading control. When intensity ratios of Aurora-A to GAPDH expression were analyzed in tumor and healthy tissues, 42 of 47 patients with CLL (89.4%) had higher levels of Aurora-A expression than had control subjects (P<0.001, Fischer's exact test; Figure 1a and b).

Analysis of Aurora-A expression by western blot, immunocytochemical, and immunohistochemical analyses. (a) Western blot analysis was performed using CD19+ B-cell lysates of chronic lymphocytic leukemia (CLL) and control subjects. MCF-7 breast cancer cells were used as a positive control. The graph shows the distribution of patients along the median in terms of Aurora-A levels expressed as the ratio of Aurora-A to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (b) Immunohistochemical analysis in a case of typical CLL involving bone marrow. Aurora-A expression was both nuclear and cytoplasmic and was more prominent in the larger prolymphocytes and paraimmunoblasts than in the smaller neoplastic cells. Double-labeling immunohistochemical staining with Aurora-A (red) and the B-cell marker anti-CD20 (brown) showed coexpression of Aurora-A in the CD20+ neoplastic B-CLL cells (inset). (c) Immunohistochemical analysis in a case of staging bone marrow, with a benign lymphoid aggregate used as a control for Aurora-A expression. Aurora-A was expressed in rare lymphocytes, especially in immunoblasts, whereas the smaller benign lymphoid cells were negative (inset). (d) Immunocytochemical analysis of subcellular localization of Aurora-A in CLL cells. Formalin-fixed bone marrow smears of untreated patients with CLL were used for immunocytochemical analysis. Aurora-A was expressed in both the cytoplasm and the nuclei of most of the tumor cells. (e) Immunocytochemical analysis of subcellular localization of phosphorylated Aurora-A in CLL cells. The phosphorylated form of Aurora-A and total Aurora-A were distributed similarly, both within the cytoplasm and nuclei of tumor cells. Compared with total Aurora-A, the phosphorylated form was expressed in fewer tumor cells. A benign lymphocyte, negative for phosphorylated Aurora-A, is present in the smear.
Immunohistochemical Findings
Aurora-A was detected in the tumor cells in aspirate, clot, or biopsy specimens of all patients with CLL. Both small and large CLL cells expressed Aurora-A with both a nuclear and cytoplasmic expression pattern. The larger cells, including prolymphocytes and paraimmunoblasts, showed strong nuclear expression of Aurora-A (Figure 1b). In contrast, Aurora-A expression in the benign lymphoid aggregates was restricted to rare large cells, and most small mature lymphocytes were negative (Figure 1c).
To better assess the subcellular distribution of Aurora-A in CLL cells, we performed immunocytochemical analyses using anti-Aurora-A antibody on fixed bone marrow aspirate smears from 15 patients with CLL. Aurora-A was seen in the cytoplasm and nuclei of the CLL cells (Figure 1d).
Double-labeling immunohistochemical staining with Aurora-A and the B-cell marker anti-CD20 showed coexpression of Aurora-A in the CD20+ neoplastic B-CLL cells (Figure 1b inset).
Immunocytochemical Findings
Aurora-A kinase exists in both active (phosphorylated at threonine 288) and inactive forms, and both are capable of inducing polyploidy in tumor cells.14 To determine whether the Aurora-A in CLL is active, we performed immunocytochemical staining with antibody against Aurora-A phosphorylated at threonine 288 on fixed smears from 15 CLL cases. Phosphorylated Aurora-A kinase was expressed in the cytoplasm and nuclei of the neoplastic cells in all cases (Figure 1e).
Quantitative Real-Time RT-PCR for Aurora-A mRNA Expression
After finding increased levels of Aurora-A expression in CLL at the protein level by western blot and immunohistochemical analyses, we tested for increased transcript levels of Aurora-A by quantitative real-time RT-PCR (Figure 2). One patient with CLL had elevated mRNA levels of Aurora-A in concordance with Aurora-A protein expression (Figure 2, CLL-A). However, Aurora-A mRNA levels in the neoplastic B cells of three patients with CLL were lower than the expression levels in the B cells of hematologically healthy control subjects (Figure 2, CLL-B, CLL-C, CLL-D). This was in contrast to the overexpression of Aurora-A as determined by western blot analysis, suggesting that the increased levels of Aurora-A in patients with CLL, compared with levels in patients with normal CD19+ B cells, are regulated by transcriptional and posttranscriptional mechanisms.

Aurora-A mRNA expression by quantitative real-time reverse transcriptase-PCR. Mean fold changes of AA mRNA expression in CLL patients and reference controls.
Chromosomal Aberrations in CLL and Aurora-A Expression
To determine whether an association exists between Aurora-A expression and the commonly detected chromosomal abnormalities in CLL, we performed conventional cytogenetic studies and/or FISH in samples from all 47 patients with CLL. One or more chromosomal abnormalities were detected in 28 (67%) of the 42 patients with elevated levels of Aurora-A. In 9 of 42 patients (21%), trisomy 12 was observed, and deletion of the ATM gene (del11q23) was observed in an additional 9 of 42 patients. These abnormalities are known to be associated with an accelerated clinical course in patients with CLL (Table 2). Fourteen patients with elevated levels of Aurora-A expression, however, showed a normal diploid karyotype.
Association between Aurora-A Overexpression and p53 in CLL
A recent report showed that Aurora-A phosphorylates p53 and enhances its mdm2-mediated degradation, suggesting that the overexpression of Aurora-A leads to proliferation due to the inactivation of the p53-dependent G1 postmitotic checkpoint.15
To determine the level of p53 expression, we performed immunohistochemical staining for p53 on bone marrow biopsy specimens from 40 patients with CLL that demonstrated elevated Aurora-A expression. We found nuclear positivity for p53 in 6 (15%) of these specimens (Figure 3a). No cases of 17p or p53 gene deletion were detected by conventional or FISH cytogenetic analysis, respectively.

Immunohistochemical analysis of p53, acetylated histone H3, and cyclin B1 in chronic lymphocytic leukemia (CLL). (a) CLL with p53+ tumor cells. p53 expression was primarily nuclear and predominantly limited to the larger prolymphocytes and paraimmunoblasts. (b) Typical p53− CLL. (c) Expression of acetylated histone H3 in CLL, expressed in both small and large tumor cells, showed a nuclear expression pattern. (d) Immunohistochemical analysis of cyclin B1 expression. The expression is nuclear, and occurred in small and large lymphocytes. Compared with acetylated histone H3, rare large cells showed nuclear expression of cyclin B1.
The overexpression of Aurora-A in patients with CLL suggests that the tumor cells in these patients are not in a resting state but may be engaged in cell-cycle progression. Histone H3, a key player in mitotic chromatin condensation, and thus cell-cycle progression, is a substrate of Aurora-A.16 We assessed for the presence of acetylated histone H3 expression in CLL by immunohistochemical analysis and observed its expression in CLL cells in all patients (Figure 3c). Thus, the coexpression of acetylated histone H3 in CLL cells overexpressing Aurora-A suggests that the Aurora-A in CLL cells is functional.
Recently, Obermann et al4 evaluated the relative proportion of CLL cells in different phases of the cell cycle by determining labeling indices (percentage of positive cells) of markers specific for each phase, including cyclin E (late G1 phase), cyclin A (S phase), and cyclin B1 (G2/M phase). They found that a large number of CLL cells resided in later phases of the cell cycle, suggesting that they had a proliferative potential. Cyclin B1 is subject to posttranscriptional modifications by Aurora-A in tumor cells, suggesting that both cyclin B1 and Aurora-A may be part of a common molecular pathway leading to tumorigenesis in cells with elevated levels of Aurora-A.17 We therefore assessed cyclin B1 expression by immunohistochemical analysis in all 47 CLL patients in our study. Only a small percentage of neoplastic cells (on average, 6%) expressed cyclin B1 (Figure 2d), and this expression was nuclear, occurring in both the small lymphocytes and the larger prolymphocytes and paraimmunoblasts. The percentage of cyclin B1+ cells in our study was slightly higher than that reported previously by Obermann et al.4 The overexpression of Aurora-A in our patients with CLL may, in part, contribute to the increased expression of cyclin B1.
Discussion
Traditionally, CLL has been thought to be characterized by a gradual accumulation of apoptotically defective neoplastic B lymphocytes, most of which reside in the G0/G1 phase of the cell cycle.18, 19 Many previous investigations have focused on the defective apoptotic features of the malignant cells, which seem to play an important role in disease progression and chemotherapy resistance. CLL, however, is composed not only of dormant lymphocytes but also of proliferating cells found in the proliferation centers in lymph nodes as well as in other disease sites, such as the bone marrow. This proliferative component of the disease may be important for CLL disease progression.3, 11, 20 Aurora kinases are mitotic kinases that play an important role in regulating the cell cycle, especially in the G2/M phase. They are important regulators of different events during mitosis such as centrosomal duplication, mitotic spindle assembly, chromosome segregation, and cytokinesis at the end of telophase.5, 6 Altered expression of all three Aurora kinases has been implicated in the pathogenesis of various cancers, and their overexpression induces chromosomal instability.8, 9
In view of these findings, we speculated that Aurora-A may be involved in the pathogenesis of CLL. We showed Aurora-A expression in both the cytoplasm and nuclei of CLL cells. We also showed Aurora-A overexpression by two independent methods, western blot analysis, and immunohistochemistry. In the normal physiologic state, the levels and activity of Aurora-A peak during the G2/M phase transition, specifically at the centrosomes and mitotic spindles. Previous studies that evaluated the expression of Aurora-A in tumor cells by immunohistochemical methods have shown that Aurora-A is detected in the cytoplasm of tumor cells at all stages of the cell cycle. This finding suggests that the role of Aurora-A in tumorigenesis is associated with the aberrant phosphorylation of cytoplasmic proteins at different phases of the cell cycle in addition to excessive phosphorylation of its physiologic substrates.21, 22, 23 Although both small and large lymphoid cells within the infiltrates expressed Aurora-A, expression was stronger and nuclear in the prolymphocytes and paraimmunoblasts, suggesting that overexpression of Aurora-A is in the proliferative component of CLL.
We also assessed smears of CLL using on antibody specific for the phosphorylated (functional) form of Aurora-A. Aurora-A was expressed in the neoplastic cells in all cases assessed, suggesting that Aurora-A kinase in CLL exists in its active form. Several lines of evidence indicate that both the active and inactive forms of Aurora-A contribute to cell proliferation and tumor progression.5, 8 Whether this is the case in CLL or whether the overexpression of Aurora-A is a consequence of the proliferative potential of prolymphocytes and paraimmunoblasts needs to be examined.
In contrast, RT–PCR to assess Aurora-A mRNA levels in CLL showed that Aurora-A mRNA expression was similar in CLL and hematologically healthy control subjects. These results suggest that mechanisms other than mRNA overexpression may contribute to Aurora-A overexpression in CLL.
In CLL patients, a functional p53 pathway is an important indicator of responsiveness to purine nucleoside analogues, perhaps explaining the prognostic importance of 17p/p53 abnormalities. p53 gene mutations are rare in CLL, and deletion or mutation is associated with disease progression, resistance to treatment, and reduced survival in patients with this disease.5 Immunohistochemical staining for p53 can be used as an, albeit imperfect, surrogate for mutated p53 because the mutated form has a longer half-life than wild-type p53, making its detection more likely by immunohistochemical analysis. In this study, 15% of CLL cases were p53+.
Recent studies provide evidence of an association between Aurora-A overexpression and chromosomal instability in human tumors.24, 25, 26 Up to 15% of patients with CLL show progressive disease associated with chromosomal abnormalities, including trisomy 12, p53 gene mutations, and ATM gene deletion. Of these three abnormalities, trisomy 12 is most common (15–20% of cases),27, 28, 29 which may occur alone or with deletions or translocations of chromosome 13q14 (25% of cases).1 13q14 deletions represent early clonal aberrations; most are undetectable at the cytogenetic level but can be detected with use of molecular probes for the 13q14 region in more than 50% of CLL cases.30 In this study group, conventional and/or FISH studies demonstrated one or more chromosomal aberrations in 67% of CLL patients with elevated Aurora-A expression, suggesting that expression of Aurora-A in CLL may contribute to chromosomal instability in these patients. However, 14 CLL patients with elevated Aurora-A expression had a diploid karyotype, suggesting that Aurora-A overexpression is not the only factor contributing to the genomic instability in CLL.
In summary, Aurora-A is expressed in CLL cells at higher levels than in the cells of healthy subjects and is active (functional) in at least a subset of CLL cells. In addition, the expression of acetylated histone H3 and cyclin B1 in CLL suggests that there is active transcription machinery in a subset of the neoplastic cells. These findings suggest that Aurora-A may be a potential therapeutic target in a subset of patients with CLL.
References
Cheson BD, Bennett JM, Grever M, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood 1996;87:4990–4997.
Lampert IA, Wotherspoon A, Van Noorden S, et al. High expression of CD23 in the proliferation centers of chronic lymphocytic leukemia in lymph nodes and spleen. Hum Pathol 1999;30:648–654.
Messmer BT, Messmer D, Allen SL, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest 2005;115:755–764.
Obermann EC, Went P, Tzankov A, et al. Cell cycle phase distribution analysis in chronic lymphocytic leukaemia: a significant number of cells reside in early G1-phase. J Clin Pathol 2007;60:794–797.
Meraldi P, Honda R, Nigg EA . Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002;21:483–492.
Higuchi T, Uhlmann F . Cell cycle: passenger acrobatics. Nature 2003;426:780–781.
Marumoto T, Zhang D, Saya H . Aurora-A—a guardian of poles. Nat Rev Cancer 2005;5:42–50.
Giet R, Prigent C . Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci 1999;112:3591–3601.
Terada Y, Tatsuka M, Suzuki F, et al. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J 1998;17:667–676.
Yakushijin Y, Hamada M, Yasukawa M . The expression of the aurora-A gene and its significance with tumorgenesis in non-Hodgkin's lymphoma. Leuk Lymphoma 2004;45:1741–1746.
Bueso-Ramos CE, Ferrajoli A, Medeiros LJ, et al. Aberrant morphology, proliferation, and apoptosis of B-cell chronic lymphocytic leukemia cells. Hematology (Amsterdam, Netherlands) 2004;9:279–286.
Schlette E, Bueso-Ramos C, Giles F, et al. Mature B-cell leukemias with more than 55% prolymphocytes. A heterogeneous group that includes an unusual variant of mantle cell lymphoma. Am J Clin Pathol 2001;115:571–581.
Bulman AS, Heyderman E . Alkaline phosphatase for immunocytochemical labelling: problems with endogenous enzyme activity. J Clin Pathol 1981;34:1349–1351.
Carmena M, Earnshaw WC . The cellular geography of aurora kinases. Nat Rev Mol Cell Biol 2003;4:842–854.
Lee EC, Frolov A, Li R, et al. Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res 2006;66:4996–5002.
Crosio C, Fimia GM, Loury R, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 2002;22:874–885.
Sasayama T, Marumoto T, Kunitoku N, et al. Over-expression of Aurora-A targets cytoplasmic polyadenylation element binding protein and promotes mRNA polyadenylation of Cdk1 and cyclin B1. Genes Cells 2005;10:627–638.
Korsmeyer SJ . BCL-2 gene family and the regulation of programmed cell death. Cancer Res 1999;59 (7 Suppl):1693s–1700s.
Bogner C, Schneller F, Hipp S, et al. Cycling B-CLL cells are highly susceptible to inhibition of the proteasome: involvement of p27, early D-type cyclins, Bax, and caspase-dependent and -independent pathways. Exp Hematol 2003;31:218–225.
Gritsko TM, Coppola D, Paciga JE, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res 2003;9:1420–1426.
Takahashi T, Futamura M, Yoshimi N, et al. Centrosomal kinases, HsAIRK1 and HsAIRK3, are overexpressed in primary colorectal cancers. Jpn J Cancer Res 2000;91:1007–1014.
Tanaka T, Kimura M, Matsunaga K, et al. Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res 1999;59:2041–2044.
Aguilar-Santelises M, Magnusson KP, Wiman KG, et al. Progressive B-cell chronic lymphocytic leukaemia frequently exhibits aberrant p53 expression. Int J Cancer 1994;58:474–479.
Miyoshi Y, Iwao K, Egawa C, et al. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer 2001;92:370–373.
Sen S, Zhou H, Zhang RD, et al. Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J Natl Cancer Inst 2002;94:1320–1329.
Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998;20:189–193.
Knuutila S, Elonen E, Teerenhovi L, et al. Trisomy 12 in B cells of patients with B-cell chronic lymphocytic leukemia. N Engl J Med 1986;314:865–869.
Que TH, Marco JG, Ellis J, et al. Trisomy 12 in chronic lymphocytic leukemia detected by fluorescence in situ hybridization: analysis by stage, immunophenotype, and morphology. Blood 1993;82:571–575.
Rowley JD, Testa JR . Chromosome abnormalities in malignant hematologic diseases. Adv Cancer Res 1982;36:103–148.
Kalachikov S, Migliazza A, Cayanis E, et al. Cloning and gene mapping of the chromosome 13q14 region deleted in chronic lymphocytic leukemia. Genomics 1997;42:369–377.
Acknowledgements
Parts of this work were presented at the US and Canadian Academy of Pathology Annual Meeting, San Diego, CA, USA, 25–29 March 2007. We thank Tamara Locke for her assistance in editing this paper and LaKisha Rodgers and Andrea Sedo for their assistance in formatting the figures and the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Inamdar, K., O'Brien, S., Sen, S. et al. Aurora-A kinase nuclear expression in chronic lymphocytic leukemia. Mod Pathol 21, 1428–1435 (2008). https://doi.org/10.1038/modpathol.2008.173
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.173
Keywords
This article is cited by
-
Differential expression of aurora-A kinase in T-cell lymphomas
Modern Pathology (2013)
-
Pharmacogenetic profiling of Aurora kinase B is associated with overall survival in metastatic colorectal cancer
The Pharmacogenomics Journal (2011)
