
Abstract
Cell surface mucin (cs-mucin) glycoproteins are constitutively expressed at the surface of respiratory epithelia where pathogens such as influenza A virus (IAV) gain entry into cells. Different members of the cs-mucin family each express a large and heavily glycosylated extracellular domain that towers above other receptors on the epithelial cell surface, a transmembrane domain that enables shedding of the extracellular domain, and a cytoplasmic tail capable of triggering signaling cascades. We hypothesized that IAV can interact with the terminal sialic acids presented on the extracellular domain of cs-mucins, resulting in modulation of infection efficiency. Utilizing human lung epithelial cells, we found that IAV associates with the cs-mucin MUC1 but not MUC13 or MUC16. Overexpression of MUC1 by epithelial cells or the addition of sialylated synthetic MUC1 constructs, reduced IAV infection in vitro. In addition, Muc1−/− mice infected with IAV exhibited enhanced morbidity and mortality, as well as greater inflammatory mediator responses compared to wild type mice. This study implicates the cs-mucin MUC1 as a critical and dynamic component of the innate host response that limits the severity of influenza and provides the foundation for exploration of MUC1 in resolving inflammatory disease.
Similar content being viewed by others
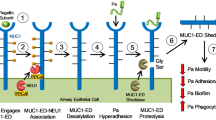


Introduction
Influenza A virus (IAV) is a highly contagious respiratory pathogen. Penetration of mucosal barriers by IAV, subsequent infection of epithelial cells, and ensuing inflammatory responses cause both acute and often severe disease. The family of mucin glycoproteins, which are highly conserved across species, have long been regarded as contributors to innate mucosal barrier function in IAV infection.1 However, studies have been limited to the secreted and gel-forming mucins2, 3, 4 with little or no attention given to the in vivo role of cell surface mucins (cs-mucins) in host defense against influenza infection.
The cs-mucins reside beneath the gel-mucin layer, being constitutively expressed at the cell surface by nearly all epithelial cells. All cs-mucin family members display large extracellular polypeptide structures supporting an array of branched and linear O-linked oligosaccharides that terminate in the sialic acid N-acetyl neuraminic acid (Neu5Ac).5 The extracellular domain of the cs-mucin glycoprotein is non-covalently linked to a transmembrane domain and can be shed from the cell surface into the lumen or internalized by clathrin-mediated endocytosis for recycling.6 These features enable cs-mucins to provide a barrier function to mucosal pathogens by displaying sites for pathogen binding, acting as a releasable decoy, and sterically blocking binding of pathogens to underlying cellular receptors. The cytoplasmic tail domain of cs-mucins is highly conserved across species, undergoes both serine and tyrosine phosphorylation, and interacts with kinases and adaptor molecules,7, 8, 9, 10, 11 consistent with a role in signal transduction in response to pathogen binding. As IAV specifically binds sialic acid-expressing receptors to trigger its entry by endocytosis into epithelial cells, the virus also has the potential to interact with sialic acid on cs-mucins, which may modulate both the infection process and the host response.
Of the cs-mucin family, the cs-mucin MUC1 (Muc1 in non-human species) has been the most extensively studied. In response to airway infection by Pseudomonas aeruginosa12 and respiratory syncytial virus,13 TLR activation in airway epithelial cells and macrophages induces the production of inflammatory mediators such as IL-8 (KC in the mouse) and TNFα to recruit effector cells.14, 15 In turn, MUC1 is upregulated and acts to suppress TLR signaling.16 In addition, production of the anti-inflammatory IL-10 and type I interferon (IFN) has been shown to correlate with increased MUC1 expression.17, 18, 19 Investigations in mice with Helicobacter pylori-induced gastritis revealed that Muc1 expression negatively regulates the NLRP3 inflammasome, which mediates production of IL-1β.20 Taken together, these data suggest that in addition to a barrier role to infection, MUC1 may also be involved with regulation of inflammation in response to infection. As over-exuberent inflammation is associated with morbidity and mortality in virulent influenza infections, we wished to explore the interaction of MUC1 with influenza virus. Using our well-established IAV infection models, we clearly show that MUC1-expressing epithelial cells have increased resistance to IAV infection and demonstrate the critical importance of MUC1 in limiting influenza-induced inflammation and death.
Results
The cell surface mucin MUC1 associates with IAV
Human lung epithelia express MUC1, MUC13 and MUC16, which have an extracellular tandem repeat domain of 2,500, 250 and 22,000 amino acids, respectively, and when fully glycosylated, extend above the cell surface to differing lengths.21 To establish whether MUC1, MUC13 and/or MUC16 can interact with IAV during the course of infection, we infected human type II alveolar epithelial (A549) cells with PR8 virus and at 1, 8, 16 and 24 h post infection (h.p.i.), labeled the cells with antibody to PR8 virus and to cs-mucin and examined the co-localization of the labeling by confocal microscopy (Figure 1a–c and Supplementary Figure S1 online). When both PR8 and the cs-mucin were visualized in the same field, it appeared there was a strong correlation of co-localization of PR8 with MUC1, as indicated by the intense orange color (Figure 1a), but not MUC13 or MUC16 (Figure 1b and c respectively).

The cs-mucin MUC1 and viral antigens co-localize on human lung epithelial cells. (a–c) A549 cells were infected with 0.1 multiplicity of infection (MOI) PR8 virus, then stained for the presence of PR8 antigens and either MUC1, MUC13 or MUC16 at 1, 8, 16 and 24 h.p.i. and visualized by confocal microscopy (See also Supplementary Figure S1). Shown is a representative image taken at 24 h.p.i. of cells, with nuclei stained with DAPI (blue), antibodies to PR8 virus (red) and the indicated cs-mucin (green). Orange indicates green and red pixel sites are co-localized. (d) Mander’s co-localization coefficients were determined using data from 5 fields of view at × 20 magnification. ***P<0.001 MUC1 compared to all other groups, ΔP<0.01 MUC16 compared to MUC13 at 16 h.p.i., two-way ANOVA. ANOVA, analysis of variance; h.p.i., hours post infection.
Using Mander’s co-localization coefficient,22 determined from 5 fields of view at × 20 magnification, we enumerated the fraction of the positively identified fluorescence pixels corresponding to PR8 virus that coincided with those for cs-mucin. We found a strong correlation of co-localization of PR8 virus with MUC1, but less so for MUC13 and MUC16 (Figure 1d). Within the first hour of infection, during which time IAV binds to sialic acid on the cell surface, we ascertained that PR8 virus was co-localized with MUC1 for ∼75% of the events, which was significantly more than for MUC13 and MUC16. At 8 h.p.i., after the virus has been internalized, the overlap coefficient of MUC1 with PR8 virus decreased to a similar level to that of both MUC13 and MUC16 (Figure 1d). At later time points, after new viral progeny are produced to infect a second round of cells, PR8 virus and MUC1 again co-localize to a significantly greater degree than PR8 virus with MUC13 and MUC16. These findings suggest there may be different functions of these three cs-mucins when expressed by A549 epithelial cells. Given the possible interaction of MUC1 with PR8 virus, we hypothesized that MUC1 may influence IAV infection of the host cell and chose this cs-mucin for further examination.
Overexpression of MUC1 at the cell surface limits IAV infection
Chinese hamster ovary-K1 (CHO-K1) epithelial cells, which do not express MUC1, can be infected by IAV but do not produce virion progeny.23 To evaluate the interaction of MUC1 with IAV during the infection process, we stably transfected CHO-K1 cells with pcDNA3.1-MUC1 to induce overexpression of this cs-mucin, which was confirmed by confocal microscopy (Figure 2a and b) and flow cytometry (Figure 2c). We then infected CHO-K1±MUC1 expressing cells with a high multiplicity of infection of PR8gfp virus. Viral entry into cells and genome transcription were assessed by detection of intracellular production of GFP, which can only occur upon successful infection of the cell. Indicating that the presence of MUC1 at the surface may limit viral entry, the CHO-K1 cells over-expressing MUC1 had a lower percentage of GFP+ cells (Figure 2d). As PR8gfp virus loses the ability to express GFP after further rounds of replication, we confirmed these findings by detection of intracellular viral nucleoprotein (NP) production during the course of infection with unmodified PR8 virus (Figure 2e). These data also showed that CHO-K1 cells expressing MUC1 were less readily infected than the control cells, which were stably transfected with the unmodified pcDNA3.1 vector and unable to express this cs-mucin.

MUC1 overexpression greatly reduces viral infection of CHO-K1 cells. Representative confocal microscopy image of (a) CHO-K1 cells, stably transfected with empty pcDNA3.1 vector, or (b) CHO-K1 cells, stably transfected with pcDNA3.1-MUC1 to overexpress MUC1. Blue = nuclei. Green = MUC1. (c) Stable MUC1 expression of the cells after at least 10 passages in selective media was confirmed by flow cytometric analysis after MUC1 staining. Histogram shows MUC1-transfected cells (blue) and control cells (red). (d) CHO-K1±MUC1 expressing cells were infected with 10 MOI PR8gfp virus and the percentage of cells producing intracellular green fluorescent protein (GFP) was determined by flow cytometry at different times post infection. (e) CHO-K1±MUC1 expressing cells were infected with 10 MOI PR8 virus and the percentage of cells producing intracellular viral NP was determined by flow cytometry. *P<0.001 unpaired T-test. MOI, multiplicity of infection.
IAV binds to MUC1 and triggers shedding of the mucin extracellular domain
To confirm a direct physical interaction between IAV and MUC1, lysates of CHO-K1 cells transfected with either MUC1 or control vector were coated onto plates and probed with PR8 virus. Antibodies specific for PR8 HA were then used to detect bound virus. As shown in Figure 3a, significantly more virus bound to plates containing lysates derived from MUC-expressing CHO-K1 cells, than control CHO-K1 cell lysates, suggesting that the additional virus had been captured by MUC1. As expected from our infection assays (Figure 2d and e), where CHO-K1 control cells were readily infected, significantly more virus bound to control cell lysates than to bovine serum albumin-coated plates due to these cells expressing a receptor for the virus.

IAV binds to MUC1 and MUC1-ED is shed after infection of cells in vitro. (a) Plates were coated with CHO-K1 cell lysates from MUC1+ and control cells, or 5% (w/v) bovine serum albumin (BSA), probed with PR8 virus, then viral antigen detected using PR8 HA-specific MAb via ELISA. Bound virus was calculated by subtracting the average OD value of a no virus control from the OD value obtained for each test well. ***P<0.001 one-way ANOVA. Data are representative of three independent experiments. (b) CHO-K1 MUC1+ and CHO-K1 control cells (n=4) were infected with PR8 virus, then the amount of MUC1-ED shed into the supernatant quantitated via ELISA. Data show a significant increase of MUC1-ED from 5–24 h.p.i. compared to uninfected CHO-K1 MUC1+ cells at time=0 (**P<0.01, ***P<0.001 one-way ANOVA). Data are representative of two independent experiments. ANOVA, analysis of variance.
As many microbial pathogens are known to trigger shedding of the MUC1 extracellular domain (ED) after binding,24, 25, 26, 27 we tested whether this phenomenon could also be caused by IAV. Supernatants were collected from PR8-infected CHO-K1 control and MUC1+ cell cultures and the amount of MUC1-ED present in the supernatant was determined by ELISA. As shown in Figure 3b, MUC1-ED levels increased as early as 3 h post-challenge with influenza and reached a plateau at 5 h.p.i. These data suggest that MUC1-ED may act as a releasable decoy by binding to IAV and upon being shed, subsequently limit infection of the underlying cell.
Synthetic MUC1 inhibits the ability of IAV to infect cells
We next evaluated the ability of synthetically generated sialylated MUC1 glycopeptides to block infection of epithelial cells. A total of four model synthetic MUC1 (sMUC1) glycopeptides were generated (Figure 4a) that represented the two simplest, and thus most synthetically accessible, O-linked sialylated glycans known as the sialyl Tn and sialyl T antigens {Neu5Ac(α2-6)GalNAc and Neu5Ac(α2-6)[Gal(β1-3)]GalNAc respectively}. Although these truncated glycans are only weakly expressed by healthy cells,28 we proposed that the presence of α2,6-linked sialic acid could mimic the decoy behavior of healthy MUC1 towards IAV. Two of the sMUC1 peptides encompassed a single copy of the 20 amino-acid MUC1 extracellular tandem repeat sequence SAPDTRPAPGSTAPPAHGVT with a single glycosylated threonine residue (Figure 5a, i and ii), while the other two glycopeptides encoded shorter sequences GSTAPPAHGVT and a glycosylated serine (Figure 5a, iii and iv). PR8 virus was incubated with equimolar amounts of each of the sMUC1 glycoproteins for 1 h at 4 °C. Incubation at 4 °C enabled attachment of the virion HA to the Neu5Ac-bound sMUC1, without the influence of viral neuraminidase activity, which can cleave Neu5Ac at higher temperatures. The co-mixtures were then tested for infectious virus by plaque formation in MDCK cells to determine whether sMUC1 could neutralize the virus. Interestingly, co-incubation with as little as 1 nmol ml−1 of the two longer 20 amino-acid sMUC1 glycopeptides resulted in a significant reduction the number of plaques formed compared with the untreated control (Figure 4b). Neutralisation appeared to be independent of whether the peptides presented sialyl-Tn or sialyl-T antigen. The shorter sMUC1 glycopeptides were unable to block infectivity of PR8 virus even when co-incubated with 50 nmol ml−1 sMUC1 glycopeptide (Figure 4c). A commercially prepared Fc-tagged MUC1 glycopeptide, which was not sialylated, was also examined for neutralization of influenza infection and found not to inhibit PR8 infection of MDCK cells (Supplementary Figure S2).

Synthetic MUC1 glycopeptides can inhibit viral infection in vitro. (a) Schematic representation of four synthetic MUC1 (sMUC1) glycopeptides: (i) and (ii) sMUC1 glycopeptides encompassed a single copy of the 20 amino-acid MUC1 extracellular tandem repeat sequence SAPDTRPAPGSTAPPAHGVT with a single glycosylated threonine residue, while (iii) and (iv) sMUC1 encoded shorter sequences GSTAPPAHGVT and a glycosylated serine. PR8 virus was incubated with either (b) 1 nmol ml−1 sMUC1, or (c) 50 nmol ml−1 sMUC1, then the remaining infectious virus quantitated by plaque assay in MDCK cells. The reduction in plaque numbers in the sMUC1:PR8 virus mixtures are represented as the percentage of the total number of plaques formed by untreated PR8 virus that underwent the same reaction conditions in the absence of sMUC1. (**P<0.01, ***P<0.001, ****P<0.0001 compared to untreated controls and compared to sMUC1:PR8 mixtures involving the shorter sMUC1 (iii and iv), one-way ANOVA with Dunnett’s multiple comparison test). ANOVA, analysis of variance.

Muc1 deficit confers a poorer outcome in response to IAV infection. Anesthetized wild type (WT) and Muc1−/− mice (mixed sex, 8–12 weeks old) were intranasally infected with 30 plaque forming unit (PFU) PR8 virus (n=10) or 500 PFU Cal7 virus (n=5), monitored for weight loss and illness, then killed if the pre-determined humane endpoint was reached. (a) Comparative survival plots. Infected Muc1−/− mice showed significantly reduced survival compared to WT mice (*P=0.0115 for PR8 virus and *P=0.0344 for Cal7 virus, Log rank Mantel-Cox text). (b) Comparison of weight loss expressed as percentage of starting weight. *P<0.05, unpaired T-test. (c) Clinical score comparison shows Muc1−/− mice had increased overt signs of illness compared to WT mice. (b) and (c) + indicates time of killing of an individual Muc1−/− mouse, Φ indicates time of killing of an individual WT mouse.
Muc1 expression reduces severity of IAV infection in vivo
Collectively, our in vitro data reveal that MUC1 and IAV interact and MUC1 expression at the cell surface may provide a mechanism that limits IAV infection. We next sought to determine whether Muc1−/− mice, which are unable to express Muc1, were more or less vulnerable to influenza infection than wild type (WT) mice (Figure 5). Groups of WT and Muc1−/− mice were infected with 30 plaque-forming units (PFU) of PR8 virus, which is a dose of virus known to be sublethal in WT mice, and assessed weight loss, morbidity and lung viral load. To determine whether the findings were generalizable to influenza viruses relevant to human infection we also tested A/California/07/09 (Cal7) virus, an H1N1 IAV strain isolated from humans during the 2009 influenza pandemic. In both cases, Muc1−/− but not WT mice reached the humane endpoint from day 6 onwards and needed to be killed as a result of their infection (Figure 5a). For PR8 virus, a sublethal infection of WT mice was converted to an infection that caused 50% lethality in mice unable to express Muc1. The Cal7 virus at a dose of 500 PFU was virulent and did result in killing of one of five WT mice but the Muc1−/− mice showed much greater disease severity with only one mouse surviving beyond day 7. In addition, both PR8- and Cal7-infected Muc1−/− mice lost substantially more weight during the acute infection (Figure 5b) and had increased overt signs of illness (Figure 5c) compared to WT mice.
Muc1 expression impedes early IAV infection in vivo
Analysis of pulmonary viral loads in mice infected with either 30 PFU PR8 virus or 100 PFU Cal7 virus (both sublethal doses in WT mice) revealed that the rate of increase of virus appeared faster in Muc1−/− mice than in WT mice over the first 2–3 days post infection (d.p.i.) (Figure 6). For PR8 virus, an approximately 20-fold greater (P<0.001, Student’s unpaired T-test) lung virus titer was observed in the Muc1−/− mice compared to WT mice on day 3 post infection. (Figure 6a). Peak viral titers were similar in both groups of mice but were delayed in WT mice by up to 2 days (Figure 6a). For Cal7 virus (Figure 5b), the significant difference in viral loads (approximately 10-fold) was seen at 2 d.p.i. and pulmonary viral loads in Muc1−/− reached peak titers 24 h earlier than in WT mice. Both Muc1−/− mice and WT infected mice appeared equally capable of clearing the infection, irrespective of the infecting virus strain, with Muc1−/− mice having significantly reduced pulmonary viral loads at 7 d.p.i. compared to the peak of infection at 3 d.p.i. These results indicate that expression of Muc1 may serve as an early physical barrier, reducing the initial rate of amplification of pulmonary virus and delaying the time to maximal viral burden.

Muc1−/− mice display a more rapid rise in pulmonary IAV loads. Groups of wild type (WT) and Muc1−/− mice (mixed sex, 8–12 weeks old) were intranasally infected with 30 plaque-forming unit (PFU) PR8 virus (n=5) or 100 PFU Cal7 virus (n=4–7, combined data from 2 experiments). Viral loads were analyzed by plaque assay of homogenized lung tissue over the course of infection. Significantly higher titers were observed in PR8-infected Muc1−/− mice at 3 d.p.i. (***P<0.001 unpaired T-test) and in Cal7-infected Muc1−/− mice at 2 d.p.i. (***P<0.0001 unpaired T-test) compared to the respective infected WT mice. Significant reduction of viral load for Muc1−/− mice at 7 d.p.i. compared to 3 d.p.i. shows evidence for clearing of virus from the lungs (ΔP=0.0002 and P=0.0001 for PR8 and Cal7 infection respectively, unpaired T-test).
IAV-infected Muc1−/− mice exhibit enhanced early inflammatory responses to infection.
Despite the initial differences in viral progression in the WT and Muc1−/− mice, the viral loads were essentially the same beyond 3 d.p.i. Nevertheless, the clinical scores remained higher throughout the course of infection in Muc1−/− mice. As excessive pulmonary inflammation is a hallmark of severe influenza disease,29 we performed a histological examination of the lungs at 3 d.p.i. (Figure 7) and investigated the inflammatory mediators and cellular recruitment to the site of infection in the two groups of mice (Figure 8). Compared to WT mice, microscopic examination of histological sections of the lungs of Muc1−/− mice infected with 30 PFU PR8 revealed a noticeably greater number of inflammatory cells in the interstitium and alveoli, most markedly surrounding the bronchioles (Figure 7a and b). To ascertain whether this enhanced pathophysiology was due to a deficiency in Muc1 regulation of inflammation, rather than as a result of heightened viral load, we next examined sections of lung taken from mice infected with 100 PFU Cal7 at 3 d.p.i., when viral titers in the lungs were equivalent (Figure 6). Indicative of a lack of an effective ability to control excessive inflammation, we observed a more diffuse lymphocytic infiltration into the alveolar spaces of Muc1−/− mice, while WT exhibited more focal sites of perivascular infiltration (Figure 7c and d). Furthermore, the more intense purple hue of the hematoxylin and eosin stained Muc1−/− tissue is suggestive of greater acidification of the lung micro-environment due to the possible increased presence of pro-inflammatory mediators.30, 31

Enhanced histopathologic changes occur in the lungs of Muc1−/− mice infected with PR8 or Cal7. Wild type (WT) (a,c) and Muc1−/− (b,d) mice were intranasally infected with 30 PFU PR8 virus (a,b) or 100 PFU Cal7 virus (c,d). At 3 d.p.i. the lungs were removed, sectioned and stained with hematoxylin and eosin for histological examination. Images (taken at × 10 magnification) show increased cellular infiltrate surrounding the bronchiole spaces in PR8-infected Muc1−/− mice (b) compared to WT mice (a). Cal7-infected wild-type mice (c) exhibited smaller foci of increased perivascular leukocyte infiltrates in the alveoli and interstitium compared to a more diffuse lymphocytic infiltration in Cal7-infected Muc1−/− mice (d). To demonstrate Muc1−/− mice exhibit normal pathology until challenged, an example of uninfected WT and Muc1−/− lungs is given in Supplementary Figure S5.

Inflammatory cells and mediators are enhanced in Muc1 deficient mice after infection with PR8 virus. WT and Muc1−/− mice (n=5, female, 8–12 weeks old) were infected with 30 PFU PR8 virus and BAL samples taken at the indicated time points after infection. BAL cellular content was then assayed for (a) CD45+ leukocytes, (b) neutrophils and (c) alveolar macrophages and for (d) IL-6, (e) MCP and (f) TNFα by CBA analysis. (*P<0.01 unpaired T-test).
To quantitate the inflammatory responses to infection, WT and Muc1−/− mice were challenged with 30 PFU PR8 virus and bronchoalveolar lavage (BAL) samples were taken at different days post infection for analysis of cellular, chemokine and cytokine content as previously described.31, 32, 33 Overall, the number of CD45+ leukocytes present in the BAL was significantly higher in Muc1−/− mice for the duration of infection (Figure 8a). On day 3 post infection, when viral loads were 20-fold higher in PR8-infected Muc1−/− mice than WT mice (Figure 6), an increase in the presence of neutrophils, and macrophages was also observed (Figure 8b and c, respectively). Consistent with increased cellular infiltrate in the airways, we found the BAL fluid from Muc1−/− mice contained significantly greater amounts of the pro-inflammatory cytokines IL-6, MCP and TNFα than WT mice at the day 3 time point and for TNFα, at subsequent time points as well (Figure 8d–f, respectively). Although levels of MIP1α, KC, MIP1β and RANTES were identical between infected mouse groups throughout the course of infection (Supplementary Figure S3), when considered together, Muc1−/− mice had heightened levels of total cytokine and chemokine content in the BAL fluid compared to WT mice, consistent with a “cytokine storm”, which may have contributed to morbidity and mortality. While we cannot rule out the possibility that the enhanced viral load in PR8-infected Muc1−/− mice and the resulting increased cellular infiltrate in the lungs at 3 d.p.i. (Figures 7b and 8a–c) contributed to the greater inflammatory cytokine response, the pulmonary viral loads at 5 and 7 d.p.i. were equivalent in WT and Muc1−/− mice. As such, the time during which Muc1−/− mice exhibited enhanced weight loss and clinical symptoms (5–8 d.p.i., Figure 5b and c, PR8 infection) was most likely induced by the increased production of inflammatory cytokines and not a result of viral load.
To further explore the viral load-dependence on cytokine secretion, we examined the BAL of Cal7 virus-infected WT and Muc1−/− mice at 3 d.p.i., a time at which viral loads were equivalent (as in Figure 6, Cal7 infection), regardless of the dose administered in the initial inoculum (data not shown for doses other than 100 PFU Cal7 virus). In contrast to PR8-infected mice, the numbers of total leukocytes, alveolar macrophages and neutrophils recruited to the BAL were not significantly different between the two groups of mice at 3 d.p.i. (Figure 9a and Figure S4). Nevertheless, consistent with a role for Muc1 in regulating inflammatory responses to infection, Cal7-infected Muc1−/− mice consistently showed significantly higher levels of cytokines and chemokines including IL-6, TNFα, MCP and RANTES compared to WT mice (Figure 9b and Supplementary Figure S4). Taken together with the histopathological observations, the inflammatory state of Muc1−/− mice infected with Cal7 appears to be more dysregulated than WT mice and would have significantly contributed to the observed increase in disease burden.

Muc1−/− mice infected with Cal7 secrete heightened inflammatory cytokines into the BAL. WT and Muc1−/− mice (n=5, female, 8–10 weeks old) were infected with 100 plaque-forming units (PFU) Cal7 virus and BAL samples taken at 3 d.p.i. (a) BAL cellular content was assayed for CD45+ leukocytes, neutrophils and alveolar macrophages by flow cytometry. (b) BAL content was assayed for IL-6, MCP-1, RANTES, IL-1a, IL-10, KC, MIP-1a, MIP-1b and TNF via cytometric bead array and flow cytometry (*P<0.05, **P<0.01, unpaired T-test).
Discussion
Our findings reveal that the cs-mucin MUC1 plays a dynamic functional role in the host-innate defences against influenza virus infection. We demonstrate IAV closely associates with MUC1 during the infection process and that sialylated MUC1 has the potential to bind to virions and reduce their ability to infect host cells. By displaying sialic acid for IAV adhesion and consequently trapping the virus at this site, MUC1 may limit binding of the virion to other sialic acid-expressing glycoproteins in the glycocalyx that potentially act as entry receptors for mediating IAV infection. Furthermore, we contend that upon interaction of IAV with the extracellular domain of MUC1, this subunit is released, extruding the virion into the lumen, while the cytoplasmic subunit is involved in signaling pathways that may lead to regulation of inflammatory mediators.34 Further supporting a role for MUC1 in innate host-defences against infection, our in vivo infection studies revealed that Muc1−/− mice were more vulnerable than WT mice to disease caused by the laboratory mouse adapted PR8 IAV and a 2009 H1N1 pandemic human IAV isolate, presumably resulting from increased viral burden early after infection and dysregulated inflammatory responses.
MUC1 is expressed by virtually all of the surface columnar epithelial cells of the respiratory tract, as well as type II pneumocytes in the alveoli,35 a major target of IAV infection.29, 36 During the normal cs-mucin recycling process in these cells, MUC1, MUC4 and MUC16 extracellular domains are shuttled to the cell surface in exosomes and upon release of the vesicle contents, will re-attach to the transmembrane domain via a SEA module, enabling continued and renewable expression of these glycoproteins at the cell surface.37 Importantly, the content of exosomes obtained from bronchoepithelial samples have been demonstrated to have a neutralizing effect on human IAV.38 Our data now provide clear in vitro and in vivo experimental evidence for a role of MUC1 as an initial physical barrier to IAV infection that may slow the early spread of the virus within the lungs. Host cell attachment by IAV is mediated by the HA glycoprotein, which binds to specific types of Neu5Ac-containing receptors.39 The towering structure of the MUC1 extracellular domain, estimated to be 200–500 nm in length when fully glycosylated,21 plus expression of an abundance of terminally linked Neu5Ac is a likely first point of contact for HA binding by IAV that has penetrated the overlying gel mucus layer and gained access to the epithelial cell surface. Such chemoattraction and adherence to specific mucin carbohydrates have also been demonstrated for mouse adenovirus type I (MAVS-1)40 and respiratory syncytial virus (RSV),13 as well as bacteria including Campylobacter jejuni,24 Helicobacter pylori26 and Pseudomonas aeruginosa.25
In this study we have been able to block IAV infection of MDCK cells with synthetic MUC1 glycopeptides expressing α2-6-linked Neu5Ac, with those having a longer peptide chain of 20 amino acids being much more efficient than glycopeptides of 11 amino acids in length. This difference may reflect steric hindrance of receptor-binding sites on adjacent monomers of the trimeric HA molecule. This function is reminiscent of the naturally occurring γ-inhibitors of IAV, such as surfactant protein A41 and PTX3,42 which inhibit IAV infection by blocking HA receptor-binding function. It will be interesting to compare these synthetic inhibitors with the natural inhibitors in vitro and in vivo.
Infection of epithelial cells induces modulation of membrane associated O-linked glycoproteins, Neu5Ac residues and heightened expression of MUC1 at the cell surface.43 Given the normal clathrin-mediated endocytosis pathway utilized for recycling extracellular MUC1 (6ref. 6) and that a major route of infection by IAV is via incorporation of the virion into the endosome after attachment to a Neu5Ac-containing receptor, we found it surprising that MUC1 did not facilitate IAV infection of host epithelial cells. This is particularly relevant as the extracellular domain of cs-mucins is endocytosed upon de-sialylation of O-glycans attached to the polypeptide backbone,6 and cleavage of sialic acid may be mediated by the active neuraminidase (NA) of bound virus. One explanation is that IAV binding to MUC1 may trigger release of the extracellular domain prior to its internalization. This “releasable decoy” mechanism is similar to what we have previously proposed for protection against C. jejuni infection in the gastrointestinal tract24 and our experiments indicating MUC1 is shed into the culture supernatant of PR8-infected CHO-K1 MUC1+ cells would support this function for influenza virus as well.
The strength of the host-innate immunological responses to IAV infection is a key determinant in clinical outcomes. The expression of MUC1 in the airways has been linked with an anti-inflammatory role through inhibition of Toll-like receptor (TLR) signaling.12 Evidence is accumulating that suggests pathogen interaction with TLRs on alveolar macrophages triggers release of TNFα, which upregulates MUC1 production by airway epithelial cells. Pathogen stimulation of the epithelial cells drives transforming growth factor-α (TGFα) release. TGFα binds to and activates the epidermal growth factor receptor (EGFR), which in turn associates with the MUC1 cytoplasmic tail leading to MUC1 phosphorylation and subsequent binding to the TLR. This results in inhibition of TLR signaling and a dampening of airway inflammation. This pathway has been elucidated in TLR5-dependent activation of primary epithelial cells in Pseudomonas aeruginosa infection,14, 15 but is also consistent with findings of negative regulation of other TLR molecules12 and with the anti-inflammatory effect of MUC1 during respiratory syncytial virus infection13 and Haemophilus influenzae infection44 of lung epithelial cells in vitro.
Other cs-mucins can also modulate inflammation showing either pro- or anti-inflammatory functions in different situations. For example, in response to TLR ligands, Muc13 in gastric mucosa enhances epithelial cell inflammatory cell signaling, the opposing function of Muc1 in the same tissue.34 However, experiments with mice lacking Muc13 show this cs-mucin protects against intestinal inflammation in the colitis model, by inhibiting toxin-induced colonic epithelial cell apoptosis.45, 46 Loss of Muc16 caused upregulation of IL-6 in the mouse cornea affecting epithelium and stroma homeostasis.47 Muc4, upregulated in nasal polyps in response to inflammation in patients with chronic rhinosinusitis, impairs the anti-inflammatory effects of corticosteroid treatment, again a contrasting role to that of Muc1 expressed in the same tissue.48 Overall, the cs-mucins appear to have critical roles in regulating inflammatory pathways in the mucosa with Muc1 clearly providing a dampening signal, supported by the data presented here.
Our study demonstrates for the first time that Muc1−/− mice are more vulnerable to IAV infection and have elevated levels of pro-inflammatory cytokines, particularly IL-6 and MCP-1, which are produced by cellular response mechanisms that are TLR dependent.49 Therefore, it is likely that in the context of IAV infection, MUC1 expression acts at two levels to defend against infection and its consequences. Initially, the large extracellular domain, which is heavily O-glycosylated with oligosaccharide structures terminating in Neu5Ac, acts as a virus trap and a releasable decoy ligand for IAV, resulting in partial dampening of infection during the early stages. Second, we contend that, as observed for other pathogens,24, 25, 26 MUC1 regulates influenza virus-induced inflammation that would otherwise contribute to more severe pulmonary disease and potentially death. Understanding the MUC1 regulatory mechanisms in the context of IAV infection is of obvious interest to our future studies and may lead to an understanding of the mechanism as to why IAV infection is as a major trigger for disease exacerbations in patients with chronic respiratory illnesses where cs-mucins are aberrantly expressed.5
Methods
Viruses. The H1N1 subtype influenza viruses A/Puerto Rico/8/34 (PR8) and A/California/7/09 (Cal7) were used in this study. PR8 is a human virus that has been adapted to show high virulence in mice, whereas Cal7 is a human isolate of the 2009 pandemic strain, which grows in mouse lungs without adaptation. PR8gfp is a reverse engineered PR8 virus that encodes green fluorescent protein (GFP) within the NS gene segment.50 Viruses were amplified in eggs and the allantoic fluid was assayed for infectious virus by plaque formation in Madin Darby canine kidney (MDCK) cells51 and expressed as PFU per ml.
Cell lines and viral infections. Chinese hamster ovary-K1 (CHO-K1) cells, MDCK cells and human type II alveolar epithelial (A549) cells were grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and antibiotics (all cell lines were originally obtained from the American Type Culture Collection). Cell cultures were maintained at 37 °C in a 5% CO2 incubator. To infect cells with IAV, confluent monolayers were washed once with phosphate-buffered saline (PBS) then infected with a known amount of infectious virus in media without FBS (infection media). The inoculum was removed after 1 h and replaced with infection media containing 1 μg ml−1 TPCK-trypsin. Cell culture supernatants were harvested at various times post infection as specified and the infectious viral titer was determined by plaque assay.51 To enumerate infected cells, monolayers were collected at the time points indicated, permeabilized using the BD Cytofix/Cytoperm Plus kit as per manufacturer’s specifications (BD Biosciences, North Ryde, NSW, Australia), then viral nucleoprotein (NP) detected by flow cytometry after staining with the mouse monoclonal antibody (MAb) NP-FITC clone D67J (Pierce, Thermo Fisher Scientific, Scoresby, Vic, Australia). Data were acquired using FACSCanto II and analyzed with FlowJo X version 10 (Tree Star, Ashland, OR). To evaluate whether Fc-tagged MUC1 and synthetic MUC1 (sMUC1) constructs limited IAV infection, virus was incubated with each of the constructs for 1 h at 4 °C in infection media. The infectious virus remaining in the co-mixtures and untreated control were then assayed by plaque formation in MDCK cells.51
MUC1 transfection. To manipulate MUC1 expression by CHO-K1 cells, a pcDNA3 vector (Invitrogen, Thermo Fisher Scientific) expressing MUC1 cDNA containing 22 variable number tandem repeats (VNTR) or the vector alone were transfected into the cells via electroporation using a Gene Pulser II (Bio-Rad, Hercules, CA), with settings of 0.246 kV and 950 μF per transfection reaction (6 × 106 cells per ml and 25 μg ml−1 plasmid DNA). Transfected cells were cultured in the presence of 1 mg ml−1 of the selective antibiotic G418 (United Bioresearch Products, Dural, NSW, Australia). Clones with high surface expression of MUC1 were obtained via limiting dilution and selected by flow cytometric analysis after staining with fluorescently conjugated mouse anti-MUC1 MAb BC2.52
Confocal microscopy. Cells were grown on coverslips and exposed to virus at a known multiplicity of infection (MOI). At the time points indicated, cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.1% Triton X-100, then treated with iFix signal enhancer (ThermoFisher Scientific). Virus was detected with sheep anti-PR8 polyclonal antiserum containing antibodies specific for PR8 virus surface antigens (BioCSL, Parkville, Vic., Australia), followed by anti-sheep Ig conjugated with Alexa Fluor (AF) 547 (Invitrogen). MUC1 was detected with BC2 MAb conjugated to AF488 (in-house). MUC13 was detected with rabbit anti-human MUC13 polyclonal Ab (Abcam, Melbourne, Vic, Australia), and AF488 anti-rabbit IgG (Invitrogen). MUC16 was detected using mouse anti-human MUC16 [OC125] (Abcam) and AF488-conjugated anti-mouse Ig (Invitrogen). Coverslips were then fixed to glass slides using ProLong Gold with DAPI (Invitrogen), and examined with an LSM 700 confocal microscope (Zeiss, North Ryde, NSW, Australia) using Zen 2010 software.
MUC1 capture ELISA. To investigate direct binding of PR8 virus to MUC1, we modified the direct microbial ELISA on plate bound mucins protocol.53 Briefly, 106 CHO-K1 control and CHO-K1 MUC1+ cells were collected and lysed for 15 min on ice in 200 μl radioimmunoprecipitation assay (RIPA) buffer containing cOmplete protease inhibitor cocktail according to the manufacturer’s instructions (Roche, Dee Why, NSW, Australia). Debris was pelleted and a 1/5,000 dilution of the resulting supernatant, or 5% bovine serum albumin in PBS was used to coat the wells of a 96-well Maxisorp Nunc Immunoplate (ThermoScientific, Waltham, MA). After overnight incubation at 4 °C, the plates were washed 3 times with PBS containing 0.05% Tween-20, then blocked with 0.5%(w/v) casein in PBS containing 0.05% Tween-20 for 1 h at room temperature. After washing, wells were incubated with 7 × 106 PFU per 50 μl PR8 virus in PBS for 1 h. The plates were washed, then virus detected with a mouse monoclonal antibody (MAb) against the PR8 HA (MAb E2.1, produced in-house) diluted in 0.5%(w/v) casein in PBS containing 0.05% Tween-20 (1 h at room temperature), washed, then HRP-conjugated rabbit anti-mouse Immunoglobulin (Ig; DAKO, North Sydney, NSW, Australia) added and incubated for a further 1 h. After a final wash step, SureBlue TMB microwell peroxidase substrate (KPL, Australian Biorearch, Balcatta, WA, Australia) was added and after 30 min and the colorimetric change stopped with a 1 M HCl solution. The OD was then read at 450 nm with wavelength correction at 570 nm. Bound virus was then calculated by subtracting the average OD value of a “no virus” control (n=4) from the OD value obtained for each test well.
MUC1 shedding ELISA. CHO-K1 control and CHO-K1 MUC1+ cells were seeded at in a 24-well plate and incubated at 37 °C, 5% CO2 overnight, with the monolayer reaching approximately 80% confluency. The culture medium was then removed and cell monolayer washed 3 times with infection media. PR8 virus (2 × 106 PFU in 200 μl infection media, approximate MOI=1) or infection media alone (as a negative control) was added to each well. At the times indicated, the supernatants from the cultures were harvested (n=4 per time point) and depleted of cells by centrifugation at 400 g for 3 min, then stored at −20 °C. To quantitate the amount of shed MUC1-ED present, we utilized the detection of mucins in solution immunoassay.54 Briefly, 100 μl of the collected supernatants and serial dilutions of RIPA lysates from 106 MUC1-expressing CHO-K1 cells (to create a standard curve), were used to coat a 96-well immunosorp ELISA plate, overnight at 4 °C. The plates were then blocked with 5% bovine serum albumin in PBS and MUC1-ED detected using a mouse 0.2 μg ml−1 BC2 MAb (which is specific for the extracellular core protein domain),52 then detected using HRP-conjugated rabbit anti-mouse Ig and SureBlue as described above. The OD was then read at 450 nm with wavelength correction at 570 nm. The amount of MUC1 in the lysate used to generate the standard curve was expressed as arbitrary units where 106 cells express a million units. The relative amount of shed MUC1-ED present in culture supernatants were determined from the standard curve generated.
Ethics statement. All experimental procedures were approved under project number 1212560 by the University of Melbourne Animal Ethics Committee (AEC) under relevant institutional guidelines, the Prevention of Cruelty to Animals Act 1986 and associated regulations, and the Australian Code for the Care and Use of Animals for Scientific Purposes 2014.
Animal model. Age and sex matched eight to twelve-week-old male or female C57BL/6 (wild type) and Muc1−/− mice on a C57BL/6 background were maintained in the Specific Pathogen Free (SPF) Physical Containment Level 2 (PC2) Bioresources Facility at the Department of Microbiology and Immunology, The University of Melbourne. All experimental inoculation procedures were conducted under general anesthesia with inhaled isofluorane at 2.5% (Baxter Healthcare Corporation). Virus was diluted in sterile PBS and administered intranasally in 50 μl to anesthetized mice. Mice were monitored daily for weight loss and disease signs and killed if a previously determined humane endpoint was reached.
Histology of mouse lungs. Lungs were removed immediately following killing, sufflated and fixed in 10% neutral buffered formalin overnight. The lungs were processed routinely, embedded in paraffin, cut into 5 μm sections and stained with hematoxylin and eosin. Slides were then scanned electronically using a Mirax Digital Slide Scanner (Zeiss) and the images examined for histopathologic alterations utilizing CaseViewer (v2.1, 3D Histech, Budapest, Hungary). All captured images were taken at × 10 digital magnification.
Assessment of the cellular and cytokine composition of bronchoalveolar lavage (BAL). BAL was extracted as previously described.31, 32, 33 Briefly, leukocytes (CD45+), neutrophils (Ly6G+, CD45+ and F480−) and alveolar macrophages (F480+, Ly6G− MHC class IIint) were enumerated by flow cytometry and expressed as a proportion of cellular events analyzed. The total numbers of each cell population was calculated in relation to the number of white blood cells per ml (WBC per ml) in the original sample, determined by counting cells with the aid of a light microscope and hemocytometer using the Trypan Blue exclusion method.
Cytometric bead array. The CBA flex set (BD Bioscience) was used as per manufacturer’s instructions to determine the cytokine concentrations in the BAL supernatant. Data were acquired using a FACSCanto II and analyzed with FlowJo X version 10 (Tree Star). Cytokine concentrations were determined with reference to standard curves using GraphPad Prism 6.0F software (La Jolla, CA).
Production of sialylated synthetic MUC1 glycopeptide. The sMUC1 glycopeptides were synthesized by Fmoc-strategy solid phase peptide synthesis using pre-prepared glycosylserine and threonine building blocks, followed by purification by reversed phase HPLC, according to the procedure reported by Corcilius and Payne.55
Statistical analysis. Statistical analyses were performed using GraphPad Prism Version 6. All graphs show mean±s.e. and are representative of at least two independent experiments.
References
Linden, S.K., Sutton, P., Karlsson, N.G., Korolik, V. & McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 1, 183–197 (2008).
Ehre, C. et al. Overexpressing mouse model demonstrates the protective role of Muc5ac in the lungs. Proc. Natl Acad. Sci. USA 109, 16528–16533 (2012).
Mata, M., Morcillo, E., Gimeno, C. & Cortijo, J. N-acetyl-L-cysteine (NAC) inhibit mucin synthesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Biochem. Pharmacol. 82, 548–555 (2011).
Buchweitz, J.P., Harkema, J.R. & Kaminski, N.E. Time-dependent airway epithelial and inflammatory cell responses induced by influenza virus A/PR/8/34 in C57BL/6 mice. Toxicol. Pathol. 35, 424–435 (2007).
Rose, M.C. & Voynow, J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 86, 245–278 (2006).
Altschuler, Y. et al. Clathrin-mediated endocytosis of MUC1 is modulated by its glycosylation state. Mol. Biol. Cell 11, 819–831 (2000).
Mukherjee, P., Tinder, T.L., Basu, G.D. & Gendler, S.J. MUC1 (CD227) interacts with lck tyrosine kinase in Jurkat lymphoma cells and normal T cells. J. Leukoc. Biol. 77, 90–99 (2005).
Lillehoj, E.P., Kim, H., Chun, E.Y. & Kim, K.C. Pseudomonas aeruginosa stimulates phosphorylation of the airway epithelial membrane glycoprotein Muc1 and activates MAP kinase. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L809–L815 (2004).
Wang, H., Lillehoj, E.P. & Kim, K.C. MUC1 tyrosine phosphorylation activates the extracellular signal-regulated kinase. Biochem. Biophys. Res. Commun. 321, 448–454 (2004).
Meerzaman, D., Shapiro, P.S. & Kim, K.C. Involvement of the MAP kinase ERK2 in MUC1 mucin signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L86–L91 (2001).
Rahn, J.J., Shen, Q., Mah, B.K. & Hugh, J.C. MUC1 initiates a calcium signal after ligation by intercellular adhesion molecule-1. J. Biol. Chem. 279, 29386–29390 (2004).
Ueno, K. et al. MUC1 mucin is a negative regulator of toll-like receptor signaling. Am. J. Respir. Cell Mol. Biol. 38, 263–268 (2008).
Li, Y., Dinwiddie, D.L., Harrod, K.S., Jiang, Y. & Kim, K.C. Anti-inflammatory effect of MUC1 during respiratory syncytial virus infection of lung epithelial cells in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L558–L563 (2010).
Kato, K., Lillehoj, E.P. & Kim, K.C. Pseudomonas aeruginosa stimulates tyrosine phosphorylation of and TLR5 association with the MUC1 cytoplasmic tail through EGFR activation. Inflamm. Res. 65, 225–233 (2016).
Kato, K. et al. Membrane-tethered MUC1 mucin is phosphorylated by epidermal growth factor receptor in airway epithelial cells and associates with TLR5 to inhibit recruitment of MyD88. J. Immunol. 188, 2014–2022 (2012).
Kim, K.C. & Lillehoj, E.P. MUC1 mucin: a peacemaker in the lung. Am. J. Respir. Cell Mol. Biol. 39, 644–647 (2008).
Yen, J.H., Xu, S., Park, Y.S., Ganea, D. & Kim, K.C. Higher susceptibility to experimental autoimmune encephalomyelitis in Muc1-deficient mice is associated with increased Th1/Th17 responses. Brain Behav. Immun. 29, 70–81 (2013).
Gaemers, I.C., Vos, H.L., Volders, H.H., van der Valk, S.W. & Hilkens, J. A stat-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J. Biol. Chem. 276, 6191–6199 (2001).
Kato, K., Lillehoj, E.P. & Kim, K.C. MUC1 regulates epithelial inflammation and apoptosis by PolyI:C through inhibition of Toll/IL-1 receptor-domain-containing adapter-inducing IFN-beta (TRIF) recruitment to Toll-like receptor 3. Am. J. Respir. Cell Mol. Biol. 51, 446–454 (2014).
Ng, G.Z. et al. The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 65, 1087–1099 (2015).
Hattrup, C.L. & Gendler, S.J. Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 70, 431–457 (2008).
Manders, E., Verbeek, F. & Aten, J. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169, 375–382 (1993).
Kumari, K., Gulati, S., Smith, D.F., Gulati, U., Cummings, R.D. & Air, G.M. Receptor binding specificity of recent human H3N2 influenza viruses. Virol. J. 4, 42 (2007).
McAuley, J.L. et al. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J. Clin. Invest. 117, 2313–2324 (2007).
Lillehoj, E.P. et al. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 280, L181–L187 (2001).
Linden, S.K. et al. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 5, e1000617 (2009).
Arcasoy, S.M. et al. MUC1 and other sialoglycoconjugates inhibit adenovirus-mediated gene transfer to epithelial cells. Am. J. Respir. Cell Mol. Biol. 17, 422–435 (1997).
Kim, Y.S., Gum, J. Jr & Brockhausen, I. Mucin glycoproteins in neoplasia. Glycoconj. J. 13, 693–707 (1996).
Guarner, J. & Falcon-Escobedo, R. Comparison of the pathology caused by H1N1, H5N1, and H3N2 influenza viruses. Arch. Med. Res. 40, 655–661 (2009).
Bordon, J. et al. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. Int. J. Infect. Dis. 17, e76–e83 (2013).
McAuley, J.L., Chipuk, J.E., Boyd, K.L., Van De Velde, N., Green, D.R. & McCullers, J.A. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 6, e1001014 (2010).
McAuley, J.L. et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2, 240–249 (2007).
McAuley, J.L. et al. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 9, e1003392 (2013).
Sheng, Y.H. et al. MUC1 and MUC13 differentially regulate epithelial inflammation in response to inflammatory and infectious stimuli. Mucosal Immunol. 6, 557–568 (2012).
Umehara, T., Kato, K., Park, Y.S., Lillehoj, E.P., Kawauchi, H. & Kim, K.C. Prevention of lung injury by Muc1 mucin in a mouse model of repetitive Pseudomonas aeruginosa infection. Inflamm. Res. 61, 1013–1020 (2012).
Ibricevic, A. et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 80, 7469–7480 (2006).
Kim, K.C. Role of epithelial mucins during airway infection. Pulm. Pharmacol. Ther. 25, 415–419 (2012).
Kesimer, M. et al. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J. 23, 1858–1868 (2009).
Gamblin, S.J. & Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 285, 28403–28409 (2010).
Nguyen, Y., Procario, M.C., Ashley, S.L., O’Neal, W.K., Pickles, R.J. & Weinberg, J.B. Limited effects of Muc1 deficiency on mouse adenovirus type 1 respiratory infection. Virus Res. 160, 351–359 (2011).
Benne, C.A. et al. Interactions of surfactant protein A with influenza A viruses: binding and neutralization. J. Infect. Dis. 171, 335–341 (1995).
Reading, P.C. et al. Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J. Immunol. 180, 3391–3398 (2008).
Lillehoj, E.P. et al. NEU1 Sialidase Regulates Membrane-tethered Mucin (MUC1) Ectodomain Adhesiveness for Pseudomonas aeruginosa and Decoy Receptor Release. J. Biol. Chem. 290, 18316–18331 (2015).
Kyo, Y. et al. Antiinflammatory role of MUC1 mucin during infection with nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol. 46, 149–156 (2012).
Sheng, Y.H. et al. MUC13 protects colorectal cancer cells from death by activating the NF-kappaB pathway and is a potential therapeutic target. Oncogene 36, 700–713 (2016).
Sheng, Y.H. et al. The MUC13 cell-surface mucin protects against intestinal inflammation by inhibiting epithelial cell apoptosis. Gut 60, 1661–1670 (2011).
Shirai, K. et al. Effects of the loss of conjunctival Muc16 on corneal epithelium and stroma in mice. Invest. Ophthalmol. Vis. Sci. 55, 3626–3637 (2014).
Milara, J., Morell, A., Ballester, B., Armengot, M., Morcillo, E. & Cortijo, J. MUC4 impairs the anti-inflammatory effects of corticosteroids in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. (2016) (in press).
Shoemaker, J.E. et al. An ultrasensitive mechanism regulates influenza virus-induced inflammation. PLoS Pathog. 11, e1004856 (2015).
Tan, H.X. et al. Recombinant influenza virus expressing HIV-1 p24 capsid protein induces mucosal HIV-specific CD8 T-cell responses. Vaccine 34, 1172–1179 (2016).
Szretter, K.J., Balish, A.L. & Katz, J.M. Influenza: propagation, quantification, and storage. Curr. Protoc. Microbiol. Chapter 15, Unit 15G 11 (2006).
Xing, P.X., Prenzoska, J. & McKenzie, I.F. Epitope mapping of anti-breast and anti-ovarian mucin monoclonal antibodies. Mol. Immunol. 29, 641–650 (1992).
Sheng, Y.H., Hasnain, S.Z., Png, C.W., McGuckin, M.A. & Linden, S.K. Techniques for assessment of interactions of mucins with microbes and parasites in vitro and in vivo. Methods Mol. Biol. 842, 297–312 (2012).
Harrop, C.A., Thornton, D.J. & McGuckin, M.A. Detecting, visualising, and quantifying mucins. Methods Mol. Biol. 842, 49–66 (2012).
Corcilius, L. & Payne, R.J. Stereoselective synthesis of sialylated tumor-associated glycosylamino acids. Org. Lett. 15, 5794–5797 (2013).
Acknowledgements
J.M. is funded by the National Health and Medical Research Council of Australia (NHMRC) Project Grant 1079924; H.X.T. receives the Australian Postgraduate Award for PhD salary support. L.B. receives funding from NHMRC Program Grant 1071916. R.P. acknowledges the Australian Research Council for Discovery Project funding (DP120100194). L.C. also acknowledges John A. Lamberton Scholarship for PhD funding. M.M. is supported by a Senior Research Fellowships from the NHMRC. We acknowledge St Jude Children’s Research Hospital, (TN, USA) as the original source of the PR8 plasmids used to reverse engineer and generate the IAV used in this study. We also acknowledge the Biological Optical Microscopy Platform at the University of Melbourne for use and maintenance of the confocal microscopy suite.
Author contributions
J.M. designed and performed the experiments, wrote the manuscript and is equal contributing senior author. H.X.T. assisted with performing the experiments. L.C. and R.P. created and provided the sMUC1 constructs and assisted with intellectual input towards data interpretation. M.M. provided the CHO-K1 cell lines and Muc1−/− mice and contributed intellectually to experimental design and data interpretation. L.B. contributed significantly to experimental design, intellectual input and manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Supplementary Material is linked to the online version of the paper
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
McAuley, J., Corcilius, L., Tan, HX. et al. The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol 10, 1581–1593 (2017). https://doi.org/10.1038/mi.2017.16
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2017.16
This article is cited by
-
Effects of particulate matter exposure on the expression of the SARS-CoV-2 ACE2 receptor in ocular surface tissues and cells
Environmental Science and Pollution Research (2024)
-
Role of the cAMP-PKA-NF-κB pathway in Mucin1 over-expression in A549 cells during Respiratory syncytial virus infection
BMC Infectious Diseases (2023)
-
Membrane curvature regulates the spatial distribution of bulky glycoproteins
Nature Communications (2022)
-
An overview of influenza A virus genes, protein functions, and replication cycle highlighting important updates
Virus Genes (2022)
-
Membrane attachment and fusion of HIV-1, influenza A, and SARS-CoV-2: resolving the mechanisms with biophysical methods
Biophysical Reviews (2022)
