
Abstract
Loss-of-function variants within the gene locus encoding protein tyrosine phosphatase non-receptor type 2 (PTPN2) are associated with increased risk for Crohn’s disease (CD). A disturbed regulation of T helper (Th) cell responses causing loss of tolerance against self- or commensal-derived antigens and an altered intestinal microbiota plays a pivotal role in CD pathogenesis. Loss of PTPN2 in the T-cell compartment causes enhanced induction of Th1 and Th17 cells, but impaired induction of regulatory T cells (Tregs) in several mouse colitis models, namely acute and chronic dextran sodium sulfate colitis, and T-cell transfer colitis models. This results in increased susceptibility to intestinal inflammation and intestinal dysbiosis which is comparable with that observed in CD patients. We detected inflammatory infiltrates in liver, kidney, and skin and elevated autoantibody levels indicating systemic loss of tolerance in PTPN2-deficient animals. CD patients featuring a loss-of-function PTPN2 variant exhibit enhanced Th1 and Th17 cell, but reduced Treg markers when compared with PTPN2 wild-type patients in serum and intestinal tissue samples. Our data demonstrate that dysfunction of PTPN2 results in aberrant T-cell differentiation and intestinal dysbiosis similar to those observed in human CD. Our findings indicate a novel and crucial role for PTPN2 in chronic intestinal inflammation.
Similar content being viewed by others


INTRODUCTION
Regulation of T helper (Th) cell differentiation into effector T-cell populations is of crucial importance to maintain tolerance towards self-antigens as well as harmless non-self antigens like those derived from food particles or commensal bacteria in the intestine and on the skin.1, 2 In Crohn’s disease (CD), a form of chronic intestinal inflammation, elevated levels of the classical Th1-cytokine IFN-γ as well as of the Th17-cytokines IL-17A/F and IL-22 have been described in intestinal biopsies and serum,3, 4, 5, 6 suggesting a misbalanced Th-cell proliferation as the disease-driving mechanism. By secretion of specific cytokines, Th cells orchestrate the immune response (e.g., class switch from IgM to IgG/IgA7 in intestinal B cells or enhanced secretion of antimicrobial peptides from intestinal epithelial cells) to commensal and pathogenic bacteria, thereby influencing the composition of the intestinal flora. Alterations in microbiota composition have been associated with intestinal disease.8
Genome-wide association studies revealed that variants within the gene locus encoding protein tyrosine phosphatase non-receptor type 2 (PTPN2) are linked to chronic inflammatory and autoimmune disorders, including rheumatoid arthritis, type 1-diabetes, celiac disease,9 and CD.10, 11, 12, 13, 14, 15 All of these diseases exhibit changes in T helper (Th)-cell proportions.16, 17 PTPN2 is ubiquitously expressed (e.g., intestinal and renal epithelium, fibroblasts, hepatocytes), but an extraordinary high expression is observed in lymphoid cells,13 including T cells. The importance of PTPN2 in immune regulation is highlighted by the fact that PTPN2-deficient mice suffer from severe inflammation and die within 2–3 weeks after birth.18 PTPN2 regulates positive selection in the thymus and controls lymphopenia-induced proliferation in CD8+ T cells.19, 20 However, it is unknown whether the loss of PTPN2 in T cells also affects CD4+ Th-cell populations during inflammation. Here, we demonstrate that the loss of PTPN2 in T cells influences Th-cell development, shifting the immune response towards a disease-promoting Th-cell composition with an increase in Th1- and Th17-cell populations but reduced regulatory functions similar as observed in CD patients. This also results in spontaneous inflammatory infiltrates into liver, kidney, and skin as well as the development of auto-reactive antibodies, pointing towards a pro- and auto-inflammatory phenotype within the whole body. These observations crucially suggest a fundamental role for genetically caused dysfunction of PTPN2 in the pathogenesis of a broad number of chronic inflammatory and autoimmune disorders.
RESULTS
T-cell-specific loss of PTPN2 aggravates intestinal inflammation
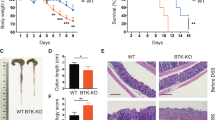
We generated mice with loss of PTPN2 in the T-cell compartment (PTPN2fl/flxCD4Cre mice) by crossing B6/C57 mice with PTPN2 allele 3 flanked by loxP sequences (PTPN2fl/fl mice; EUCOMM) with mice expressing Cre recombinase under control of the CD4 promoter (CD4Cre mice21). Specific loss of PTPN2 in T cells was confirmed by immunofluorescent staining and FACS analysis (Supplementary Figure 1 online and data not shown), whereas loss of PTPN2 in CD4+ dendritic cells was incomplete (Supplementary Figure 1). To address whether the loss of PTPN2 in T cells results in altered susceptibility for T-cell-driven intestinal inflammation, we used the transfer colitis model. Naive CD4+ cells, isolated either from PTPN2fl/fl or PTPN2fl/flxCD4Cre mice, were injected intraperitoneally (i.p.) into Rag2-deficient (Rag2−/−) mice. Although injection of PTPN2 competent naïve T cells resulted in the onset of colitis symptoms starting 4 weeks after injection, transfer of PTPN2-deficient T cells resulted in much earlier disease onset as observed by severe weight loss and bloody diarrhea starting already at week 2 post injection (Figure 1a). At week 5 after T-cell transfer, mice receiving PTPN2-deficient T cells showed enhanced macroscopic colitis score, increased spleen weight, enhanced myelin peroxidase activity and pronounced shortening of the colon when compared with mice receiving PTPN2-competent T cells (Figures 1b and c). All of these observations are indicative for a more severe intestinal inflammation. These findings were also reflected in histological assessment of mouse colon, where enhanced infiltration and pronounced epithelial damage was observed in mice receiving PTPN2-deficient T cells (Figure 1d).

Transfer of PTPN2-deficient T cells results in aggravated colitis. RAG2−/− mice were injected i.p, with PBS, 2.5 × 105 naïve CD4+ T cells from PTPN2fl/fl (WT) or PTPN2fl/flxCD4Cre (KO) mice (n=8 per group). Shown are (a) weight development post injection; (b) representative pictures and assessment of colitis severity according to the MEICS of colonoscopy performed at week 5 post injection; (c) Spleen weight, MPO activity and colon length 5 weeks post injection; each dot represents a single mouse. (d) Representative pictures show H&E staining of the terminal colon. The graph below summarizes scoring for epithelial damage and inflammatory infiltration. (e) Chronic colitis was induced in PTPN2fl/fl and PTPN2fl/flxCD4Cre mice and spleen weight analyzed (left); each dot represents a single mouse. Representative pictures from spleens of non-treated 9-month-old PTPN2fl/fl and PTPN2fl/flxCD4Cre littermates are shown. Asterisks indicate significant differences (*=P<0.05, **=P<0.01, ***=P<0.001). H&E, hematoxylin and eosin; MEICS, murine endoscopic index of colitis severity; MPO, myelin-peroxidase; PTPN2, protein tyrosine phosphatase non-receptor type 2.
To address the role of PTPN2 in T cells during colonic inflammation in a setting where a normal adaptive intestinal immune system is developed, we used a model of dextran sodium sulfate (DSS)-mediated epithelial injury. Here, food particles and intestinal microbes can penetrate the mucosal barrier and provoke an immune response. Nevertheless, as this model is mediated by epithelial erosion, it has its limitations in reflecting the complex immunological setting of human inflammatory bowel disease (IBD). The results obtained in T-cell transfer colitis were confirmed in both an acute (2.5% DSS for 7 days; Supplementary Figure 2) and a chronic DSS colitis model (four cycles consisting of 7 days 2% DSS followed by 10 days recovery phase each; Supplementary Figure 3). PTPN2fl/flxCD4Cre animals showed pronounced weight loss, aggravated shortening of the colon, enhanced myelin peroxidase activity, increased macroscopic disease activity, and pronounced immune cell infiltration upon DSS challenge. Interestingly, myelin peroxidase levels were already enhanced in PTPN2fl/flxCD4Cre mice even without DSS administration and enhanced numbers of lymphoid follicles were detected in the terminal colon of PTPN2fl/flxCD4Cre animals. Over 9-months-old PTPN2fl/flxCD4Cre mice displayed enlarged spleens even without DSS challenge (Figure 1e). Taken together, this demonstrates an increased susceptibility to acute as well as chronic intestinal inflammation upon loss of PTPN2 in T cells.
Loss of PTPN2 results in altered signal transducers and activators of transcription (STAT) phosphorylation and T-cell transcription factor expression
Th cell differentiation relies on the specific activation of STAT molecules22 and PTPN2 has been shown to influence STAT activation in myeloid cells.23 We detected enhanced levels of inflammation-induced STAT1 and STAT3 phosphorylation in colon samples from PTPN2fl/flxCD4Cre mice or Rag2−/− mice receiving PTPN2-deficient T cells, where STAT5 and STAT6 phosphorylation was reduced compared with PTPN2fl/fl mice or mice receiving PTPN2-competent T cells, respectively (Figure 2a, Supplementary Figure 4, and data not shown). These changes are likely to result directly from a T-cell intrinsic change in the response to inflammatory cytokines, as the same effects were observed in naive CD4+ T cells cultured under different Th-cell-inducing conditions.24, 25 PTPN2-deficient naïve T cells showed increased STAT1/STAT3 but reduced STAT6 phosphorylation under Th1/Th17 and Th2 conditions, respectively (Figure 2b). Interestingly, total STAT1 levels where enhanced in PTPN2-deficient T cells whereas STAT5 levels were reduced (Figure 2b) even without treatment. Consistent with enhanced STAT1/STAT3 but reduced STAT6 phosphorylation, we also found enhanced levels of T-bet (TBX21) but reduced levels of GATA-3 mRNA expression. No significant change was observed for mRNA levels of RORC and FoxP3, whereas mRNA levels of PTPN2 are clearly decreased in PTPN2-deficient mice (Figure 2c). PTPN2 expression was slightly increased in Th1, Th2, and Th17 cells (Figure 2c), possibly reflecting that PTPN2 is induced by several inflammatory cytokines.

Increased STAT1 and STAT3 phosphorylation upon loss of PTPN2. (a) RAG2−/− mice were injected i.p. with PBS, 2.5 × 105 naïve CD4+ T cells from PTPN2fl/fl (WT) or PTPN2fl/flxCD4Cre (KO) mice and killed 5 weeks post injection. Protein lysate from whole colon pieces were analyzed by western blot for phospho-STAT1 (Tyr701), total STAT1, phospho-STAT3 (Tyr705), total STAT3, phospho-STAT4 (Tyr693), total STAT4, phospho-STAT5 (Tyr694), total STAT5, phospho-STAT6 (Tyr641), total STAT6, PTPN2, and β-actin, n=8 per group, pictures show representative blots from one mouse per group. (b and c) Naïve CD4+ T cells isolated from PTPN2fl/fl (WT, empty bars) or PTPN2fl/flxCD4Cre (KO; filled bars) mice were cultured under indicated Th-cell skewing conditions for (b) 30 min, and analyzed by western blot for the same proteins as in a; or (c) 24 h and analyzed for mRNA expression of Th-cell-associated transcription factors T-bet, RORγT, FoxP3, and GATA-3, as well as PTPN2. Expression is normalized to β-actin and αCD3/αCD28-treated cells, data are representative for two independent experiments with n=3 each. KO, knockout; PBS, phosphate-buffered saline; PTPN2, protein tyrosine phosphatase non-receptor type 2; STAT, signal transducers and activators of transcription.
Increased levels of Th1 and Th17 cells in colon, MLN, and spleens of PTPN2-deficient mice
Increased infiltration of lymphoid cells and enhanced colitis severity together with elevated STAT1/STAT3 activation prompted us to address if loss of PTPN2 in T cell leads to altered Th-cell differentiation. We did not detect a difference in the proportions of CD8+ vs. CD4+ T cells between PTPN2fl/fl and PTPN2fl/flxCD4Cre mice (data not shown).
Injection of naïve CD4+ T cells into RAG−/− mice resulted in a robust induction of IFN-γ+ as well as IL-17+ cells within the intestinal CD4+ T-cell pool. While the induction of IFN-γ+ cells was moderate and IL-17+ cell induction was low in mice receiving PTPN2-competent T cells, injection of PTPN2-deficient naïve T cells resulted in massive levels of IFN-γ+ and induction of IFN-γ/IL-17 double-positive CD4+ T cells (Figure 3a). In contrast, FoxP3+/CD25high regulatory T cells (Tregs) were robustly induced in mice receiving PTPN2-competent T cells, but only mildly in mice receiving PTPN2-deficient T cells (Figure 3b).

Enhanced Th1- and Th17-cell numbers but reduced Treg induction in mice lacking PTPN2 in T cells. RAG2−/− mice were injected i.p. with PBS, 2.5 × 105 naïve CD4+ T cells from PTPN2fl/fl (WT) or PTPN2fl/flxCD4Cre (KO) mice and killed 5 weeks post injection. Lymphocytes isolated from LPL, mLN and spleen were (a) treated for 6 h with ionomycin/PMA in the presence of Brefeldin A before staining for IL-17 and IFN-γ expression or (b) directly stained for CD25 and FoxP3 expression. Representative data from one single mouse per group; gating strategy is depicted in Supplementary Figure 5a. (c) mRNA was isolated from colon tissue and analyzed for expression of the indicated Th-cell-associated Transcription factors and cytokines. (d) Serum levels of indicated cytokines from the same mice. Each dot represents a single mouse; asterisks denote statistical significance (*=P<0.05, **=P<0.01). LPL, lamina propria; PMA, phorbol myristate acetate; PTPN2, protein tyrosine phosphatase non-receptor type 2.
One limitation of the acute DSS model is the less pronounced induction of pathogenic T cells when compared with other colitis models. Nevertheless, the changes in Th-cell proportions as observed in the transfer colitis, were reflected in both DSS colitis models. We found increased levels of IFN-γ+CD4+ T cells in PTPN2fl/flxCD4Cre animals even in absence of DSS challenge, but reduced induction of CD25high/FoxP3+ cells upon inflammatory insult (Supplementary Figure 5). Consistent with there observations, we found increased mRNA levels of the Th1- and Th17-associated transcription factors T-bet and RORC, respectively, and enhanced levels of IFN-γ transcription in whole colon extracts from mice with PTPN2-deficient T cells in all three colitis models (Figure 3c, Supplementary Figure 6a and data not shown). Inflammation-induced increase in mRNA levels of FoxP3, IL-10, and IL-13 was reduced in samples from mice harboring PTPN2-deficient T cells (Figure 3c, Supplementary Figure 6b and data not shown). Inflammation-induced IFN-γ TNFα, IL-17, and IL-6 serum levels were enhanced, but IL-10 serum levels were reduced in mice harboring PTPN2-deficient T cells (Figure 3d, Supplementary Figure 6c).
The observed changes in Th-cell development are not restricted to the colonic lamina propria, as we found similar changes in IFN-γ+ and IFN-γ+/IL17+ CD4+ and Treg populations in MLN and spleens from either PTPN2fl/flxCD4Cre animals or Rag2−/− mice receiving PTPN2-deficient T cells (Figure 3a, Supplementary Figure 7a+b). These observations point towards the induction of a pro-inflammatory T-cell phenotype upon PTPN2 dysfunction promoting inflammation in the individual.
Loss of PTPN2 in T cells leads to T-lymphocyte infiltration in extra-intestinal organs and autoimmune reactions
The enhanced disease-promoting potential of PTPN2-deficient T cells is not restricted to the intestine. Administration of four cycles of DSS treatment or injection of naïve T cells resulted in perivascular lymphoid cell infiltration into the liver (Figure 4a, Supplementary Figure 8). In PTPN2fl/flxCD4Cre mice, intrahepatic infiltration was already observed in non-treated animals, which was enhanced in animals receiving DSS (Figure 4a). Injection of PTPN2-deficient T cells resulted in drastically enhanced liver infiltrates (Supplementary Figure 8). A large portion of the infiltrates in the livers of PTPN2fl/flxCD4Cre mice consisted of CD3+ cells (not shown), but other inflammatory cells, including granulocytes and other mononuclear cells, were also present. In approximately 25% of DSS-treated PTPN2fl/flxCD4Cre mice, we detected lymphatic cell infiltrations in the kidneys (Figure 4b). Of the older (>9 months) PTPN2fl/flxCD4Cre mice, 10% suffered from spontaneous skin lesions and hematoxylin and eosin staining from ear sections showed inflammatory infiltrates in the skin of these animals (Figure 4c). Of note, we found circulating anti-dsDNA, anti-nuclear, and anti-mitochondrial antibodies in PTPN2fl/flxCD4Cre mice as well as increased levels of circulating IgG and IgA antibodies, which were further increased upon DSS challenge (Figures 4d and e). These observations strongly point towards enhanced systemic inflammation and autoimmune reactions in animals being PTPN2-deficient in T cells and suggest a crucial role for PTPN2 in preventing chronic inflammation and autoimmune diseases.

Spontaneous inflammatory infiltrates in kidney, liver, and skin upon loss of PTPN2. PTPN2fl/fl or PTPN2fl/flxCD4Cre animals were treated for four cycles with 2% DSS, and representative pictures from H&E-stained sections show inflammatory infiltrates into (a) liver and (b) kidney. (c) Pictures of H&E-stained ear sections of non-treated, over 9-month-old PTPN2 PTPN2fl/fl and PTPN2fl/flxCD4Cre mice suffering from spontaneous skin lesions. (d and e): Serum from PTPN2fl/fl and PTPN2fl/flxCD4Cre animals receiving four cycles of 2% DSS were analyzed for (d) anti-dsDNA, ANA, and AMA) antibodies and (e) total IgG and IgA levels. Data are representative results from two independent experiments with at least n=6 mice per group, each. Dots represent individual mice, asterisks denote statistical differences (*=P<0.05, **=P<0.01). AMA, anti-mitochondrial antibody; ANA, anti-nuclear antibody; DSS, dextran sodium sulfate; H&E, hematoxylin and eosin; PTPN2, protein tyrosine-phosphatase non-receptor type 2.
Loss of PTPN2 induces intestinal dysbiosis
Intestinal dysbiosis is a key feature of CD26 and Th cells are involved in the regulation of antimicrobial responses. Therefore, we addressed whether the profound difference in Th-cell composition in PTPN2fl/flxCD4Cre mice results in alterations in intestinal microbiota composition. Pyrosequencing of 16S DNA and subsequent analysis with Silva and Genegreens database revealed that DSS-induced intestinal dysbiosis, as characterized by an increase of Bacteroidetes and Proteobacteria with a concomitant decrease in Firmicutes, was pronounced in PTPN2fl/flxCD4Cre animals and mild dysbiosis was already detectable in non-challenged PTPN2fl/flxCD4Cre mice (Figure 5a). A similar intestinal dysbiosis is observed in CD patients,27 although some conflicting reports exist whether Bacteroidetes are increased or decreased in human IBD.8, 27 On a family level, the decrease in Firmicutes resulted from decreased abundance of Lactobacillaceae and Lachnospiraceae while the increase in Proteobacteria was due to increased levels of Helicobacteraceae and Enterobacteriaceae (Figure 5b). DSS-induced effects were pronounced in PTPN2fl/flxCD4Cre animals. At the genus level, and especially in DSS-treated PTPN2fl/flxCD4Cre mice, we did not only find an increase in the opportunistic and potentially pathogenic Escherichia-Shigella, Flexispira (Helicobacter), and Erwinia, but also a marked decrease in Coprococcus (Figures 5c and d).

Loss of PTPN2 in T cells results in intestinal dysbiosis. Acute DSS colitis was induced in PTPN2fl/fl and PTPN2fl/flxCD4Cre mice and cecum content analyzed for microbial composition using 16S sequencing and Silva reference database on (a) phylum, (b) family, and (c) genus level. Data represent the average composition for each group (n=6 per group). (d) Relative abundance of DSS induced or reduced bacteria. (e) The graph shows Butyryl-CoA:CoA transferase transcripts per gram cecum content normalized to non-treated PTPN2fl/fl animals. n=6 per group, depicted is the mean of each group. *=P<0.05, **=P<0.01. DSS, dextran sodium sulfate; PTPN2, protein tyrosine phosphatase non-receptor type 2.
Coprococcus are the most abundant butyrate-producing bacteria in mice. They occupy the same ecological niche and have a similar function in the murine gut ecosystem than Roseburia species or Faecalibacterium in humans. CD is associated with decreased Faecalibacterium prausnitzii abundance27, 28 and reduced overall butyrate levels.29 The decrease in predominant butyrate-producing Coprococcus prompted us to assess the abundance of butyrylCoA:acetateCoA-transferase gene (ButCoA-transferase) in cecum content in order to confirm the overall impact on the butyrate-producing community. The ButCoA-transferase gene is critically involved in the butyrate production pathway of a vast majority of butyrate-producing bacteria30 and can be used as a marker for predominant butyrate-producing species.30 DSS treatment caused a reduction of ButCoA-transferase gene abundance. In PTPN2fl/flxCD4Cre animals, ButCoA-transferase gene levels were already decreased without DSS challenge, with a further decrease upon DSS administration (Figure 5e).
This demonstrates that loss of PTPN2 in T cells affects the composition of the intestinal microbiota towards a microbiota dysbiosis pattern that is similarly as observed during human chronic intestinal inflammation, e.g., CD. During inflammation, intestinal dysbiosis seems to be aggravated in PTPN2fl/flxCD4Cre mice and might contribute to the overall enhanced colitis severity in these animals. However, because a limited number of six mice per group were analyzed, further studies would strengthen our findings.
Presence of the CD-associated PTPN2 variant results in elevated Th1- and Th17-associated gene expression in intestinal biopsies from IBD patients
The CD-associated SNP rs1893217, resulting in a T>C conversion in an intron within the gene locus encoding PTPN2, leads to a malfunctional PTPN2 protein transcript.31 To address a possible role of this PTPN2 variant in T-cell development in human disease, intestinal samples from non-IBD control patients being homozygous for the wild-type (T) allele and CD patients either being homozygous for the WT (T) or the CD-associated variant (C) allele were collected from non-inflamed and inflamed regions. We detected an increase in TBX21, FoxP3, IFNG, IL-17, IL-13, and IL-10 mRNA in biopsies from inflamed regions of patients carrying the WT allele, when compared with control patients. Interestingly, T-bet, IFN-γ, and IL-17 mRNA levels were further enhanced in patients being homozygous for the CD-associated PTPN2 variant, whereas the increase in IL-13 and IL-10 was no longer detected (Figures 6a and b). PTPN2 transcription levels were enhanced in biopsies from inflamed regions in all CD patients independent of the allele variant (Supplementary Figure 9). Presence of the CD-associated PTPN2 variant led to increased serum levels of IFN-γ and IL-17 when compared with CD patients carrying the PTPN2 WT allele, whereas serum levels of IL-10 and IL-13 were not significantly altered (Figure 6c). These findings mirror our results obtained in the colitis mouse models, supporting the notion that presence of the C allele in the gene locus encoding PTPN2 leads to a loss of function of the protein transcript.31 Our observations indicate that presence of the C variant in CD patients crucially affects T-cell homeostasis and promotes T-cell alterations that are correlating to those observed in human CD.

Enhanced expression of Th1- and Th17-associated transcription factors/cytokines in inflamed regions of CD patients with the CD-associated PTPN2 variant. (a and b) Intestinal biopsies from control (Ctr) patients (n=15), or non-inflamed and inflamed regions of CD patients homozygous either for the PTPN2 WT allele (PTPN2 TT, n=8) or the CD-associated PTPN2 allele (PTPN2 CC, n=8), were analyzed for the expression of (a) TBX21, IFNG, RORC, and IL17; as well as (b) GATA3, FOXP3, IL13, and IL10. (c) Serum samples from the same patients as in (a and b) were analyzed for IFN-γ, IL-17, IL-13, and IL-10 levels. Asterisks denote significant differences (*=P<0.05, **=P<0.01). CD, Crohn’s disease; IBD, inflammatory bowel disease; n.d.=not detected; PTPN2, protein tyrosine phosphatase non-receptor type 2.
DISCUSSION
Our results demonstrate that the loss of PTPN2 in T cells influences Th-cell development, shifting the immune reaction more towards a disease-driving response, promoting inflammation and reducing regulatory functions. These findings are commonly observed during chronic inflammatory and autoimmune human diseases. In the intestine, where myriads of bacteria are present, the proper balance between inflammatory and regulatory T cells is of crucial importance to maintain tolerance towards commensals and food antigens while still conferring efficient protection against pathogenic bacteria. To date, a role for PTPN2 in T-cell development had only been described for the CD8+ cell subset.19 Here, we demonstrate a completely novel role for PTPN2 in regulating the differentiation of CD4+ Th cell subsets. Loss of PTPN2 resulted in increased levels of Th1- and Th17 cells but reduced levels of Treg cells in the intestine, MLN, and spleen. Consistent with enhanced levels of Th1- and Th17-cell populations being proposed as important drivers of intestinal pathology,32 this shift away from Treg and towards Th1 and Th17 cells resulted in enhanced intestinal inflammation in mice. In line with PTPN2 being associated with many inflammatory diseases, inflammatory infiltrates observed in several organs, including liver, kidney, and skin point towards a pro-inflammatory phenotype within the whole body.
The changes observed in mice match the situation in CD patients homozygous for the malfunctional SNP rs1893217 C variant. In these patients, we detected increased levels of Th1- and Th17-associated genes, but reduced regulatory and Th2-associated gene transcripts in biopsies from inflamed regions. In patient samples, however, changes in Th17-associated genes/cytokines were more pronounced than in mice. PTPN2 plays an important role in epithelial cell integrity,33 and cytokine secretion from antigen-presenting cells (APCs);31, 34 hence, the more pronounced Th17 phenotype in patients might reflect malfunctional PTPN2 not only in T cells but also in all cell types present in the intestine. We found increased levels of IL-13 and IL-10 expression in WT CD patients. These cytokines are associated with increased levels of Th2 and Treg cells, cell subsets not regarded as drivers of inflammation in CD. However, intestinal inflammation as observed in CD promotes the accumulation of Th cells in general, which might explain the observed increase of these two cytokines.
The enhanced disease-promoting potential of PTPN2-deficient T cells seem not to be restricted to the intestine, as mice lacking PTPN2 in T cells display inflammatory infiltrates into liver, and later also into kidney and skin. Although being involved in several inflammatory diseases, so far no correlations have been reported between PTPN2 variants and skin disorders. With respect to CD, however, this spontaneous inflammatory infiltrates are of great interest, as extra-intestinal disease manifestations are a common complication in CD.35 However, a possible correlation between PTPN2 variants and extra-intestinal manifestations has not yet been addressed. Our data suggest that the skin and kidney lesions result from a generalized loss of tolerance upon T-cell-specific loss of PTPN2. This points out the importance of PTPN2 in regulating T-cell homeostasis with respect to several T-cell-mediated autoimmune and inflammatory disorders. As we have previously shown that the activation of PTPN2 is sufficient to ameliorate acute DSS-induced colitis in mice,36 our findings might open the avenue for a completely novel treatment option for a broad number of chronic inflammatory and autoimmune diseases, such as CD, rheumatoid arthritis, type 1-diabetes, celiac disease, psoriasis, or systemic lupus erythematosus.
In contrast to previously described PTPN2-LckCre mice that also lack PTPN2 in T cells, where a moderate increase of regulatory T cells was found,19 we detected reduced levels of Tregs. Later thymic expression of the Cre recombinase under the CD4 promoter compared with the Lck promoter37 or loss of PTPN2 in CD4+ DCs38 might contribute to this effect. However, the reduction of PTPN2 in CD4+ DC was incomplete, probably as a result of the (unknown) insert location of the CD4-Cre transgene. Nevertheless, our data provide the direct and highly important further development of the findings by Wiede et al.,15 because we actually demonstrate for the very first time a mechanistic role for PTPN2 in the pathogenesis of intestinal inflammation and T cell differentiation during colitis in vivo. In contrast to Wiede et al.15 who assessed their data in mice without any disease-promoting stimuli and in non-inflamed conditions, we analyzed our mice in the setting of an inflammatory response, i.e., during colitis. This might well explain the different results with respect to Tregs observed in both manuscripts. Our data indicate that the inflammation-driven development of induced Treg, which are generated in relatively high numbers during intestinal inflammation, seems to be disturbed upon loss of PTPN2 in T cells, whereas Wiede et al. demonstrated that the development of natural Treg, which develop in the thymus, as well as steady-state Treg generation in the periphery, is slightly enhanced. Especially in the intestine, but also in more distant organs, the microbial composition influences the immune system and the induction of Treg.39 Hence, differences in the microbial composition between different animal facilities might further contribute to the differences in Treg induction in our studies compared with the findings of Wiede et al. Further, there might be the possibility that the observed effect with respect to intestinal and systemic inflammation are, at least in part, due to alterations in APCs in PTPN2 knock-out mice compared with wild-type animals. However, this argument might only account for the conditional PTPN2fl/flxCD4Cre knock-out animals, in which some CD4-expressing APC might be PTPN2-deficient. However, in mice used for T-cell transfer colitis, the loss of PTPN2 is certainly restricted to T cells and PTPN2 is surely present in all of the APC. Therefore, a possible loss of PTPN2 in APC might only, if at all, contribute to the effects observed in the PTPN2fl/flxCD4Cre knock-out animals, but not in the effects observed in mice undergoing T-cell transfer colitis.
A further pathogenic key feature of CD is the presence of intestinal dysbiosis, and in certain animal models, where genetic alterations result in enhanced colitis severity, altered intestinal flora is observed.40, 41 However, it is unknown whether these changes are the result or cause of inflammation. Nevertheless, co-housing—and thereby transferring microbiota—of transgenic mice with WT animals resulted in elevation of colitis severity in WT animals.40 This indicates that changes in intestinal microbiota promote—or at least maintain—disease. Hence, the pronounced dysbiosis observed in PTPN2xCD4Cre animals might contribute to the pronounced colitis. Some publications indicate an increase in Bacteroidetes ssp. in IBD,27 while others demonstrate the opposite—at least in some patient groups.8 Hence, our findings in T-cell-specific PTPN2-deficient mice might not completely correlate with the situation in human IBD. However, our findings provide evidence, that genetic predisposition influences the intestinal composition and might therefore help to explain differences in the intestinal flora between patient groups. Of note are also the reduced levels of butyrate-producing bacteria. Butyrate is an important energy source for intestinal epithelial cells,42 while in activated T cells, butyrate leads to enhanced expression of the pro-apoptotic Fas receptor resulting in enhanced apoptosis.43 Reduced levels of available butyrate might affect apoptosis in effector T cells, ultimately contributing to sustained intestinal inflammation.
Taken together, we demonstrate that the loss of PTPN2 in T cells results in promotion of pathogenic T-helper cell subsets, resulting in enhanced susceptibility to intestinal inflammation. Moreover, the loss of PTPN2 results in a general loss of tolerance in older, normally not autoimmune-predisposed B6/C57 mice, resulting in inflammatory infiltrates in many organs. Our findings obtained in mice are resembled in CD patients featuring the CD-associated gene variant, and intestinal dysbiosis in our mouse models closely correlates to that observed in CD patients. These observations support a crucial role for PTPN2 in the pathogenesis of chronic inflammatory diseases and CD in particular, and suggest that dysfunction of PTPN2 could be one of the driving forces for chronic inflammation and autoimmunity in the human.
METHODS
Mice and colitis induction
CD4Cre mice on B6/C57 background were purchased from Taconic (Cologne, Germany), RAG2−/− mice from Janvier (Saint-Bellevin Cedex, France), PTPN2fl/fl from EUCOMM. Mice lacking PTPN2 in T cells were generated by crossing PTPN2fl/fl mice with CD4Cre animals. All mice uses for experiments were female and were kept in a specific pathogen-free facility and experiments were performed with approval of the local animal welfare authority, namely the Veterinarian Office of the Canton Zürich, Switzerland. For transfer colitis, naïve T cells were isolated using Miltenyi naïve T cell isolation kit II (Miltenyi, Gladbach, Germany) and 2.5 × 105 cells injected i.p. into RAG2−/− hosts. Purity of naïve T cells (defined as CD4+/CD62Lhigh/CD44low/CD25− cells) was >94% as analyzed by flow cytometry. Acute DSS colitis was induced by administration of 2.5% DSS (MP Biomedicals, Carlsbad, CA) in the drinking water for 7 days. Mice were killed at day 10. Chronic DSS colitis was induced by administration of four cycles consisting of 7 days 2% DSS followed by 10 days normal drinking water. Mice were killed 4 weeks after the last DSS cycle. For all experiments, littermate controls were used.
Assessment of colitis severity and histological score
Animals were anesthetized i.p. with 90–120 mg kg−1 body weight ketamine (Vétoquinol, Bern, Switzerland) and 8 mg kg−1 body weight Xylazine (Bayer, Lyssach, Switzerland). Animals were examined as described previously.44 Recording was performed with the Karl Storz Tele Pack Pal 20043020 (Karl Storz Endoskope, Tuttlingen, Germany). Colonoscopy was scored using the murine endoscopic index of colitis severity (MEICS) scoring system as described previously44 using the following five parameters: (i) transparency of the colon, (ii) changes of the vascular pattern, (iii) fibrin visible, (iv) granularity of the mucosal surface, and (v) stool consistency. Histological scoring for inflammatory infiltration and epithelial cell damage was performed on hematoxylin and eosin-stained section of the most distal 1 cm of the mouse colon.44, 45
In vitro Th-cell differentiation
Naïve T cells were isolated using Miltenyi naïve T cell kit II. Purity was >97% as analyzed by flow cytometry. For Th differentiation, 0.5 × 106 cells per ml were stimulated with 1 μg ml−1 plate-bound anti-CD3 and 1 μg ml−1 soluble anti-CD28 under Th1-skewing (10 ng ml−1 IL-12, 10 ng ml−1 IFN-γ plus 10 μg ml−1 anti-IL-4), Th2-skewing (10 ng ml−1 IL-4, 10 μg ml−1 anti-IFN-γ plus 10 μg ml−1 anti-IL-12), Th17-skewing (1 ng ml−1 TGFβ1, 10 ng ml−1 IL-6, 10 μg ml−1 anti-IL-4, 10 μg ml−1 anti-IFN-γ plus 10 μg ml−1 anti-IL-12), or Treg-skewing (3 ng ml−1 TGFβ1, 20 ng ml−1 IL-2, 10 μg ml−1 anti-IL-4, 10 ng ml−1 anti-IFN-γ plus 20 μg ml−1 anti-IL-12) conditions in RPMI 1640 medium (Life Technology, Carlsbad, CA) supplemented with 2% fetal calf serum, and penicillin-streptomycin (Sigma, Buchs, Switzerland).
Microbiota analysis
High-throughput sequencing was performed on cecal DNA using a 454 Life Sciences system in combination with Titanium chemistry (Roche AG, Basel, Switzerland). Sequencing and analyses were carried out at Microsynth (Balgach, Switzerland). The V3–V5 hypervariable 16S rRNA region was targeted. Amplicons were quantitated using a Quant-iT PicoGreen dsDNA assay kit (Life Technologies). Taxonomic classification for every read was performed using Mothur based on the Bayesian method,46 using a kmer size of 8 bp and 1,000 iterations. As reference databases, the Greengenes reference taxonomy (updated June, 2013)47 and Silva reference taxonomy48 were used. Bootstrap values below a cutoff of 70% were marked as unclassified.
Patient samples
Intestinal tissue specimens were taken by endoscopy from macroscopically inflamed as well as non-inflamed regions of the terminal ileum, colon, or rectum of patients with CD (wild-type (TT): n=15, age range: 27–66, mean: 47.4±13.5 years; variant (CC): n=8, age range: 28–69, mean: 43.8±18.3 years), and from non-IBD control patients (n=8, age range: 37–61, mean: 49.1±9.5). Control patients were asymptomatic and presented for colon cancer screening. Written informed consent was obtained before specimen collection and studies were approved by the local Ethics Committee. Patients were matched for age, gender, and disease activity. PTPN2 genotype for the CD-associated SNP rs1893217 was determined using TaqMan technology (Life Technologies) as described previously.31
Statistical analysis
Unless otherwise stated, data are representative for one of two independent experiments with n replicates each, data are represented as average and standard error of the mean. Statistical significances were determined using Wilcoxon-Mann Whitney test.
References
Palmer, M.T . & Weaver, C.T . Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat. Immunol. 11, 36–40 (2010).
Marwaha, A.K . et al. Th17 cells in autoimmunity and immunodeficiency: protective or pathogenic? Front. Immunol. 3, 129 (2012).
Fujino, S . et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003).
Maloy, K.J . & Powrie, F . Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474, 298–306 (2011).
Sugimoto, K . et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Brand, S . et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G827–G838 (2006).
Hirota, K . et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat. Immunol. 14, 372–379 (2013).
Frank, D.N. St et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 104, 13780–13785 (2007).
Festen, E.A . et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. 7, e1001283 (2011).
Khor, B ., Gardet, A . & Xavier, R.J . Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Barrett, J.C . et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 40, 955–962 (2008).
Todd, J.A . et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 39, 857–864 (2007).
Doody, K.M ., Bourdeau, A . & Tremblay, M.L . T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: lessons from the knockout mouse model and implications in human disease. Immunol. Rev. 228, 325–341 (2009).
Franke, A . et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42, 1118–1125 (2010).
Jostins, L . et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Ferraro, A . et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 60, 2903–2913 (2011).
Shao, S . et al. Th17 cells in type 1 diabetes. Cell. Immunol. 280, 16–21 (2012).
Heinonen, K.M . et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 103, 3457–3464 (2004).
Wiede, F . et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Invest. 121, 4758–4774 (2011).
Wiede, F ., La Gruta, N.L . & Tiganis, T . PTPN2 attenuates T-cell lymphopenia-induced proliferation. Nat. Commun. 5, 3073 (2014).
Lee, P.P . et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15, 763–774 (2001).
Zhu, J ., Yamane, H . & Paul, W.E . Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489 (2010).
Scharl, M ., Hruz, P . & McCole, D.F . Protein tyrosine phosphatase non-receptor Type 2 regulates IFN-gamma-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 16, 2055–2064 (2010).
Pappu, B.P . & Dong, C . Measurement of Interleukin-17. Current Protocols in Immunology, John Wiley & Sons, (2001).
Fitch, F.W ., Gajewski, T.F . & Hu-Li, J . Production of TH1 and TH2 Cell Lines and Clones. Current Protocols in Immunology, John Wiley & Sons, (2001).
Tamboli, C.P . et al. Dysbiosis in inflammatory bowel disease. Gut 53, 1–4 (2004).
Joossens, M . et al. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut 60, 631–637 (2011).
Machiels, K . et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283 (2013).
Wang, W . et al. Increased proportions of bifidobacterium and the lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 52, 398–406 (2014).
Louis, P . & Flint, H.J . Development of a semiquantitative degenerate real-time pcr-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl. Environ. Microbiol. 73, 2009–2012 (2007).
Scharl, M . et al. Crohn's disease-associated polymorphism within the PTPN2 gene affects muramyl-dipeptide-induced cytokine secretion and autophagy. Inflamm. Bowel Dis. 18, 900–912 (2012).
Olsen, T . et al. TH1 and TH17 interactions in untreated inflamed mucosa of inflammatory bowel disease, and their potential to mediate the inflammation. Cytokine 56, 633–640 (2011).
Scharl, M . et al. Protection of epithelial barrier function by the Crohn's disease associated gene protein tyrosine phosphatase n2. Gastroenterology 137, 2030–2040 e5 (2009).
Scharl, M . et al. Protein tyrosine phosphatase N2 regulates TNFalpha-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 60, 189–197 (2011).
Barrie, A . & Regueiro, M . Biologic therapy in the management of extraintestinal manifestations of inflammatory bowel disease. Inflamm. Bowel Dis. 13, 1424–1429 (2007).
Moron, B . et al. Activation of protein tyrosine phosphatase non-receptor type 2 by spermidine exerts anti-inflammatory effects in human THP-1 monocytes and in a mouse model of acute colitis. PLoS One 8, e73703 (2013).
Wang, X . et al. TCR-dependent transformation of mature memory phenotype T cells in mice. J. Clin. Invest. 121, 3834–3845 (2011).
Vremec, D . et al. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978–2986 (2000).
Smith, P.M . et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Couturier-Maillard, A . et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 123, 700–711 (2013).
Zenewicz, L.A . et al. IL-22 deficiency alters colonic microbiota to be transmissible and colitogenic. J. Immunol. 190, 5306–5312 (2013).
Ploger, S . et al. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. NY Acad. Sci. 1258, 52–59 (2012).
Zimmerman, M.A . et al. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G1405–G1415 (2012).
Becker, C ., Fantini, M.C . & Neurath, M.F . High resolution colonoscopy in live mice. Nat. Protoc. 1, 2900–2904 (2006).
Obermeier, F . et al. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 116, 238–245 (1999).
Wang, Q . et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
DeSantis, T.Z . et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072 (2006).
Quast, C . et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Acknowledgements
This research was supported by a grant from Fonds zur Förderung des akademischen Nachwuchses of the Zürcher Universitätsverein to M.S., a research grant from the Swiss Philanthropy Foundation to M.S. and G.R., a research credit grant from the University of Zurich to M.S., research grants from the Swiss National Science Foundation to M.S. (Grant No. 314730-146204 and No. CRSII3_154488 / 1), G.R. (Grant No. 310030-120312), S.R.V. (Grant No. 320000-114009/3 and 32473B_135694/1) and the Swiss IBD Cohort (Grant No. 3347CO-108792) and by a grant from the Zurich Center for Integrative Human Physiology of the University of Zurich to M.S. The funding institutions had no role in the study design, n the collection, analysis and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest. Microsynth as a commercial service provider was involved in sequencing/analysis of fecal DNA samples, but had no role in manuscript writing.
Additional information
Author contributions
MRS performed experiments, analyzed the data, and wrote the first draft of the manuscript; S.K., C.G., T.R., I.F., S.L., and K.A. performed experiments and were involved in data analysis; F.M. and B.B. were involved in flow cytometry analysis; C.C. and C.L. performed microbiota analysis; S.R.V., M.F., G.R., and M.S. were involved in acquisition of patient samples; M.S. conceived, designed, and supervised the study. All authors wrote, corrected, and approved the manuscript.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Spalinger, M., Kasper, S., Chassard, C. et al. PTPN2 controls differentiation of CD4+ T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol 8, 918–929 (2015). https://doi.org/10.1038/mi.2014.122
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.122
This article is cited by
-
Temperature-triggered in situ forming lipid mesophase gel for local treatment of ulcerative colitis
Nature Communications (2023)
-
A small molecule inhibitor of PTP1B and PTPN2 enhances T cell anti-tumor immunity
Nature Communications (2023)
-
Targeting protein phosphatases in cancer immunotherapy and autoimmune disorders
Nature Reviews Drug Discovery (2023)
-
Loss of protein tyrosine phosphatase non-receptor type 2 reduces IL-4-driven alternative macrophage activation
Mucosal Immunology (2022)
-
Inflammatory bowel disease: between genetics and microbiota
Molecular Biology Reports (2020)
