
Abstract
Chronic mucosal inflammation is the hallmark of important and common airway diseases, such as allergic rhinitis (AR) and asthma. Lipoxin A4 (LXA4) is an endogenous pro-resolving mediator for mucosal inflammation that decreases allergic and asthmatic responses. Lipoxin B4 (LXB4) is a structurally distinct member of the lipoxin family that signals in a manner distinct from LXA4. LXB4 is generated by mucosal tissues, but its actions in allergic inflammation are unknown. Here, we used murine models of AR and asthma to investigate LXB4’s activity in mucosal inflammation. In the upper airway, LXB4 significantly decreased nasal mucosal leukocytes and degranulation of mast cells (MCs) and eosinophils. In the lower airway, LXB4 significantly decreased airway inflammation, mucus metaplasia, and hyper-responsiveness. Inhibition of MC degranulation in vivo by LXB4 was more potent than dexamethasone, and these agents displayed unique profiles for cytokine regulation; however, their overall anti-inflammatory actions were comparable. LXB4 decreased eotaxin-dependent eosinophil chemotaxis, IgE-mediated MC degranulation, and expression of type 2 cytokine receptors. Together, these findings indicate that LXB4 carries cell type selective and mucosal protective actions that broaden the lipoxin family’s therapeutic potential for upper and lower airway catabasis.
Similar content being viewed by others


INTRODUCTION
Allergic airway diseases, such as rhinitis and asthma, are common and adversely impact patients’ function and quality of life.1, 2 Allergic rhinitis (AR) and atopic asthma can present with chronic mucosal inflammatory processes, and have been referred to as a “united airways” disease.3, 4 Despite distinct clinical manifestations, these diseases have immunological similarities. For example, eosinophil tissue infiltration and IgE-mediated mast cell (MC) activation through FcɛRI bound allergen are major factors in the pathogenesis of AR and atopic asthma.5 Current treatments for AR and asthma principally target symptom relief and anti-inflammation with glucocorticosteroids (GC). Since GC are associated with heterogeneous therapeutic responses and can have serious side effects, there is still a need for new treatment strategies to control AR and asthma. Importantly, no curative therapy is available to resolve allergic airway inflammation.
Recently, research focused on the physiological termination of inflammatory responses has led to the identification of specialized mediators involved in the natural resolution of inflammation in health, including lipoxins (i.e., lipoxin A4 (LXA4) and lipoxin B4 (LXB4)).6 These specialized pro-resolving mediators are lipoxygenase-derived eicosanoids formed from arachidonic acid by transcellular metabolism during cell–cell interactions (reviewed in Levy et al.6). Lipoxins are generated by the upper respiratory tract, including by nasal polyps,7 and are present in nasal lavage fluids from aspirin-challenged patients with aspirin-exacerbated respiratory disease.8 Lipoxins are also present in the lower airways in broncho-alveolar lavage fluid (BALF) from patients with asthma.9 Of note, severe asthma is associated with decreased LXA4 generation.9 Administration of a stable analog of LXA4 blocks allergic airway inflammation, mucus metaplasia, and hyper-reactivity to methacholine.10 LXA4 is a potent inhibitor of granulocyte recruitment and activation, including eosinophils.10 LXB4 is a structurally distinct product of arachidonic acid metabolism. LXB4 regulates neutrophil activation;11 however, its actions in allergic inflammation and on the functional responses of mucosal effector cells (i.e., MC and eosinophils), have yet to be established. Here, we provide the first evidence that LXB4 mediates anti-inflammatory and pro-resolving actions for allergic airway responses in murine models of upper and lower airway mucosal inflammation.
RESULTS
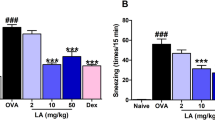
LXB4 reduces leukocyte infiltration and mucus secretion in the nasal mucosa
Mice (BALB/c) were systemically sensitized with ovalbumin (OVA)/aluminum hydroxide (Alum) and then challenged with OVA intranasally to target the allergic responses to the nasal mucosa (Figure 1a, see Methods). Twenty-four hours after the last nasal allergen challenge, mice were given LXB4 (as validated in Supplementary Figure S1 online) (100 ng, intravenous (IV)), dexamethasone (100 ng), or vehicle control for 3 consecutive days (protocol days 28, 29, and 30) (Figure 1a). Mice were killed 24 h later (protocol day 31). Histopathological assessment of the nasal cavity in mice exposed to vehicle revealed mucosal allergic inflammation with mucin secretion and leukocyte infiltration (Figure 1b). Relative to vehicle, LXB4 reduced inflammatory infiltrates and mucin secretion in the lumen (Figure 1c). Equivalent amounts of dexamethasone also decreased the mucosal inflammation, but to a lesser degree than LXB4 (Figure 1d). BALF that was collected from these mice at the time of nasal tissue excision did not reveal significant increases in eosinophils or other leukocytes (data not shown).

Lipoxin B4 (LXB4) reduces leukocyte infiltration and mucus secretion in the nasal mucosa. (a) Days 0 and 7: ovalbumin (OVA)/aluminum hydroxide (Alum) intraperitoneal (IP) sensitization, days 14–27: OVA intranasal (IN) challenge, days 29–30: vehicle (0.1%), LXB4 or dexamethasone (100 ng) intravenous (IV) injection, day 30: sacrifice. (b–d) Hematoxylin and eosin (H&E)-stained mucosal sections of BALB/c mice injected with vehicle (b), LXB4 (c), or dexamethasone (d). Red arrows—mucus; blue arrows—leukocyte infiltration sm—submucosa; mc—mucosa; b—bone ( × 20 magnification—left panel; × 40 magnification—right panel).
LXB4 has pro-resolving actions on select nasal mucosa cell populations
Leukocyte infiltration and total nasal mucosal cell numbers were significantly decreased by LXB4 compared with vehicle (Figure 2a). Differential cell count performed on Wright-Giemsa stained cytospins of nasal mucosa cells indicated a significant decrease in eosinophils numbers with LXB4 and dexamethasone (Figure 2b). The numbers of MC still containing granules significantly increased with LXB4 while dexamethasone gave no significant changes from vehicle (Figure 2c).

Lipoxin B4 (LXB4) has pro-resolving effects on select nasal mucosa cell populations. (a) Total cell number as evaluated in trypan blue exclusion test. (b and c) Differential counts of eosinophils (b) and granulated mast cells (c) in stained cytospins from BALB/c mice (n≥8), *P<0.05, **P<0.01, ***P<0.001.
MC and eosinophil degranulation is decreased by LXB4
Examination of the cytospins following Wright-Giemsa staining revealed striking evidence of changes in leukocyte granule contents (Figure 3). Mice (BALB/c) exposed to vehicle had highly degranulated eosinophils and MC with evidence for released MC granules (Figure 3a). In sharp contrast, nasal mucosal MC from LXB4-treated mice were larger and retained most of their granule contents, indicative of a less activated state (Figure 3b). Eosinophils also appeared to be less degranulated (Figure 3b). Nasal mucosal MC following dexamethasone treatment showed cellular changes consistent with partial activation and degranulation with cells generally smaller in size than with LXB4 (Figure 3c). Next, the apparent decrease in eosinophils number and degranulation was validated by flow-cytometry analysis where Siglec-F+ cells (Supplementary Figure S2a) and CD107a+ cells (Supplementary Figure S2b) were significantly decreased by LXB4 (3.50±0.87 Siglec-F+ cells; 19.54±1.74% CD107a+ cells, P<0.05) compared with vehicle (8.51±1.86% Siglec-F+ cells; 27.15±1.706% CD107a+ cells). In comparison, dexamethasone decreased Siglec-F expressing cells (4.59±0.79%, P<0.05, Supplementary Figure S2a), but had no significant effect on cell degranulation (CD107a+, P=0.531, Supplementary Figure S2b).

Fewer degranulated mast cells and eosinophils were evident in nasal mucosa with Lipoxin B4 (LXB4) treatment. (a–c) Images of Wright-Giemsa stained cytospins of vehicle (a), LXB4 (b), and dexamethasone (c) injected BALB/c mice ( × 40). Representative images from three mice. Black arrows—mast cells; hollow arrowheads—mast cell granules; and red arrows—eosinophils.
LXB4 decreases serum OVA-specific IgE levels and select cytokines/chemokines
Serum titers of OVA-specific IgE (BALB/c mice) were significantly decreased with LXB4 (0.57±0.10 pg ml−1, P<0.01) and dexamethasone (0.56±0.08 pg ml−1, P<0.01) relative to vehicle (1.54±0.29 pg ml−1) (Figure 4a). Additionally, LXB4 significantly decreased serum levels of several mediators associated with IgE production and allergic responses (Figure 4b–l), including IL-4 (Figure 4b; P<0.05), IL-9 (Figure 4c; P<0.05), RANTES (CCL5) (Figure 4d; P=0.011), granulocyte macrophage colony-stimulating factor (GM-CSF; Figure 4e; P<0.01), vascular endothelial growth factor (Figure 4f; P=0.018), and IL-12 (Figure 4h; P<0.01). Monocyte chemoattractant protein-1 was also decreased by LXB4, but did not reach statistical significance with this sample size (Figure 4g; P=0.053). Dexamethasone did not share LXB4’s broad immunoregulatory actions at this time point, but did decrease serum IL-4, IL-9, and GM-CSF (Figure 4b, c, and e). Of interest, LXB4 gave modest increases in keratinocyte chemoattractant (KC) (Figure 4i; P<0.01) and IL-13 (Figure 4j, P<0.05) levels. The increased serum KC level was not associated with increased tissue neutrophils in the nasal mucosa. Levels of IL-5 were not significantly influenced by either LXB4 or dexamethasone (Figure 4k). Serum levels of IL-17 were also detected and significantly downregulated by LXB4 (Figure 4l, P<0.01), but not by dexamethasone (Figure 4l).

Lipoxin B4 (LXB4) decreases ovalbumin (OVA)-specific IgE levels and modulates various cytokines in serum of allergic rhinitis (AR) mice. (a) OVA-specific IgE levels in vehicle, LXB4, and dexamethasone injected BALB/c mice (n=9). (b–l) Cytokine expression in serum of vehicle, LXB4, and dexamethasone injected mice (n≥5), *P<0.05; **P<0.01 two-tailed Student’s t-test.
LXB4 decreases bone marrow-derived MC degranulation and bone marrow-derived eosinophil chemotaxis
To quantitatively determine whether LXB4 carried direct actions for cell functional responses, IgE-sensitized bone marrow (BM)-derived MC (BMMC) from naive mice were exposed to LXB4 and compared with LXA4, dexamethasone, or vehicle. Cell activation for degranulation was initiated in a non-antigen-specific manner with an IgG (Fab)2′ antibody as in Lu-Kuo et al.,12 see Methods. BMMC degranulation, monitored by the percentage of β-hexosaminidase release, was significantly decreased with LXB4 (19.90±0.86% of total release relative to vehicle control cells: 39.11±0.83%, P<0.0001) (Figure 5a). LXA4 also decreased BMMC degranulation to 23.97±1.64% of total release (P<0.01), and dexamethasone (10–1,000 nM) exhibited concentration-dependent inhibition of BMMC degranulation that relative to vehicle (38.07±0.4917) was similar in magnitude to LXB4 (100 nM dexamethasone: 33.93±0.5301, P<0.01; 1,000 nM dexamethasone: 27.05±0.3910, P<0.0001) (Figure 5b). To determine whether these cellular responses were strain specific, BALB/c and FVB cell responses were determined. Both BALB/c (Supplementary Figure S3a and b) and FVB-derived (Supplementary Figure S3c and d) BMMC responded similarly to IgE-mediated activation and degranulation, which was inhibited by LXB4 and LXA4 in a concentration-dependent manner. For eosinophil responses, murine BM-derived eosinophils were exposed to LXB4, and for comparison LXA4 or vehicle, before eotaxin exposure, to determine their impact on chemotaxis (see Methods). Relative to vehicle, LXB4 and LXA4 significantly decreased eotaxin-mediated chemotaxis (Figure 5c).

Lipoxin B4 (LXB4) decreases BMMC degranulation and BM eosinophils chemotaxis. Cells were derived from BALB/c mice BM as described in Methods. (a and b) BMMC IgE-dependent activation (β-hexosaminidase release) (c) BM eosinophil chemotaxis to eotaxin presented as number of events counted per 20 s in the flow cytometer (see Methods) (n=3), **P<0.01, ***P<0.001.
LXB4 and LXA4 decrease cytokine release from BMMC following IgE-mediated activation
On the basis of the serum cytokine regulation of AR mice by LXB4, the direct actions of lipoxins on BMMC cytokine release were next determined. BMMC were exposed to LXB4, LXA4, or vehicle before activation. After 18 h, IgE-mediated activation of BMMC led to a significant TNF-α release that was significantly inhibited by LXB4 and LXA4 in a concentration-dependent manner (Figure 6a–d). LXB4 also decreased FCɛRI expression (Supplementary Figure S4a and b) without affecting cell viability (Supplementary Figure S4c).

Lipoxin B4 (LXB4) and LXA4 decrease TNF-α release from BMMC following IgE-mediated activation. BMMC were derived from BALB/c and FVB mice BM and treated as described in Methods. IgE activation was carried out with anti-rat IgG (Fab)2′ (10 μg ml−1) (a and b) TNF-α levels (pg ml−1) in BALB/c cell supernatants following incubation with LXB4 (a) and LXA4 (b). (c and d) TNF-α levels (pg ml−1) in FVB cell supernatants following incubation with LXB4 (c) and LXA4 (d), *P<0.05, **P<0.01.
LXB4 regulates allergic lung inflammation
To determine whether the actions of LXB4 in the lower airways were similar to the upper airways, we first investigated the anti-inflammatory activity of LXB4 (Figure 7I, Prevention). OVA sensitized animals (FVB) received LXB4 (100 ng, 1,000 ng) or vehicle (IV) 30 min before OVA aerosol challenge on protocol days 14–17 and were then killed 24 h later on day 18—a time of peak allergic inflammation. LXB4 led to a significant dose-dependent decrease in total BALF cell numbers (Figure 7Ia), BALF eosinophils (Figure 7Ib), and BALF lymphocyte numbers (Figure 7Id) but no significant change in macrophage numbers was seen (Figure 7Ic).

Lipoxin B4 (LXB4) regulates allergic lung inflammation. FVB mice were sensitized and aerosol challenged with ovalbumin (OVA). LXB4 (100, 1,000 ng) or vehicle was injected: (I) Prevention—30 min before challenge (days 14–17). (II) Treatment—after the last challenge (days 18–20). Total cell (a and e), eosinophil (b and f), macrophage (c and g), and lymphocyte (d and h) numbers in BALF. The resolution interval-Ri (i) for vehicle/LXB4 injected mice (n≥5), *P<0.05, **P<0.01, ***P<0.001 one-tailed Student’s t-test.
Given these anti-inflammatory actions, LXB4’s pro-resolving actions in the lower airways were next determined by administration after the allergic airways responses were fully established (Figure 7II, Treatment). Twenty-four hours after the last OVA aerosol challenge, LXB4 (100 ng, IV) was given (protocol day 18) and repeated on days 19 and 20. Mice were killed 24 h later (day 21) during the resolution phase of this model. Relative to vehicle, LXB4 significantly accelerated the resolution of the allergic lung inflammation with decreased BALF total cell number (Figure 7IIe) and decreased eosinophils, macrophage and lymphocyte numbers (Figure 7IIf–h). The resolution interval (Ri) was calculated for BALF total cells (∼6 days with vehicle) and LXB4 shortened the Ri by over 30% to ∼4 days (Figure 7IIi).
LXB4 promotes the resolution of airway hyper-responsiveness and lung inflammation
In addition to airway inflammation, airway hyper-responsiveness (AHR) is a clinical hallmark of asthma;1 therefore, the impact of LXB4 on methacholine-triggered increases in lung resistance was determined. Relative to vehicle, mice (FVB) exposed to 100 ng LXB4 treatment (as in Figure 7II) had significantly less AHR to methacholine (Figure 8a). In addition, histological analysis of lung sections from animals not subjected to AHR testing revealed that LXB4 led to significant reductions in leukocyte infiltration and epithelial cell activation (Figure 8b, c). Mice treated with LXB4 (100 ng) also exhibited a faster resolution of mucus metaplasia, as evidenced by decreased Periodic Acid Schiff-positive airway epithelial cells compared with vehicle (Figure 8d). A significant decrease in BALF Th2 cytokines (Figure 8e–g) was observed, particularly for IL-4 (Figure 8e, vehicle: 27.68±12.98; LXB4:15.51±7.40, P<0.05) and IL-13 (Figure 8g, vehicle: 3.21±1.145; LXB4: 0.845±0.483, P<0.05). Given the importance of IL-13 signaling in airway mucus metaplasia and airway hyperreactivity to methacholine,13, 14, 15, 16 we next determined the influence of LXB4 on IL-13 receptors (Figure 8h–j). Lung expression of the shared IL-4 receptor decreased during resolution (day 21) relative to the peak of inflammation (day 18), and LXB4 further accelerated this decrease in lung IL-4 receptor expression (Figure 8h). LXB4 administration also markedly decreased the expression of IL-13-specific receptors IL-13Rα1 and IL-13Rα2 (Figure 8i, j).

Lipoxin B4 (LXB4) promotes the resolution of airway hyper-responsiveness and lung inflammation. (a) Determination of airway hyper-responsiveness to metacholine, (n≥5), *P<0.05 one-tailed Student’s t-test (b and c) hematoxylin and eosin (H&E) and (d) Periodic Acid Schiff (PAS) staining of FVB mice lungs. Arrow heads point to mucus (magenta positive) Goblet cells ( × 20). (e–g) IL-4, IL-5, and IL-13 cytokine levels in BALF, (n≥5) *P<0.05 paired Student’s t-test. (h–j) mRNA levels of lung IL-4R, IL-13Rα1, and IL-13Rα2 (n≥5) *P<0.05.
DISCUSSION
AR and asthma are common allergic conditions with high morbidity. Allergen avoidance is often difficult and impractical and there is no curative therapy for the resultant allergic inflammation. Thus, new therapeutic approaches are needed. To this end, LXB4 (∼5 μg kg−1) displayed pro-resolving actions for allergic mucosal inflammation in murine models of AR and asthma. LXB4 decreased mucosal leukocyte infiltration, OVA-specific IgE, MC, and eosinophils degranulation, mucus metaplasia, and AHR to methacholine.
LXB4 mediated direct actions on pivotal cellular effectors for allergy, namely MC and eosinophils (as shown on cell functional responses in vitro), as well as indirect actions on these cells (regulation of levels of inflammatory mediators). Distinct from their important role during the induction of allergic airway responses, the levels of type 2 cytokines during resolution and their response to LXB4 varied. In resolving upper and lower airways, LXB4 potently decreased serum IL-4 and IgE levels. Because significant changes were not present in IL-5 levels, LXB4 regulation of eosinophils function appears to have resulted from a direct action on these cells as well as from the reduced levels of GM-CSF. Surprisingly, LXB4 increased serum IL-13 as the upper airways resolved. In contrast, BALF IL-13 was decreased by LXB4, suggesting local and distinct regulatory mechanisms. Although IL-13 can initiate allergic inflammation, it can also have a role in limiting inflammation. Of note, IL-13 has the capacity to induce the expression of 15-lipoxygenase, which catalyzes the formation of both LXB4 and LXA4.6
IL-4 has important roles in isotype switching and IgE production,17, 18 and LXB4 inhibited both IL-4 and serum IgE levels. Given the differential actions on IL-13 levels, expression of its receptors was determined. Lung expression of IL-4R, IL-13Rα1, and IL-13Rα2 was downregulated during resolution and further decreased by LXB4. IL-4R-deficient mice display weak Th2 responses.19 IL-13Rα1 deficiency has an established role in allergic airway responses.20 Thus, LXB4 regulation of IL-4R and IL-13Rα1 expression would serve to decrease mucus metaplasia and airway hyper-reactivity. No LXB4-mediated increases in expression of the IL-13 decoy receptor IL-13Rα2 were apparent. In asthma, regulation of IgE and IL-13 has beneficial actions in some asthmatic patients,21, 22 so together these findings suggest therapeutic potential for LXB4 or its stable analogs.23
The increase in serum KC with LXB4 was unexpected and distinct from LXA4.24 In addition to its well-defined role as a neutrophil chemoattractant, KC can modulate ciliary function of murine sinonasal epithelial cells,25, 26 suggesting that LXB4 might enhance mucociliary clearance via this mechanism. In contrast to KC, serum IL-17 levels were low and significantly decreased by LXB4. Compared with the GC dexamethasone, LXB4’s actions were of similar potency for decreasing inflammatory cell activation and more potent for regulating degranulation in vivo. Although consistently decreased by LXB4, there was donor-to-donor variability for the magnitude of stimulus-driven BMMC degranulation, suggesting context-dependent responses for MC and their regulation by LXB4. Direct cellular actions for LXB4 were also identified in vitro for inhibition of BMMC TNF-α production and BM-derived eosinophil chemotaxis. Together, these lines of evidence support potent anti-allergic and pro-resolving actions for LXB4 on MC and eosinophils that were relevant for both upper and lower airways.
Notably, LXA4 blocks histamine release from MC during interactions with epithelial cells27 and decreases neutrophil degranulation of azurophilic granules.28 In conjunction with LXB4’s actions here in vitro, the lipoxins (LXA4 and LXB4) carry MC and granulocyte stabilizing activity. Apart from its direct regulation of MC and eosinophils, LXB4 blocks T-cell activation, cytokine release,29 and neutrophil chemotaxis.11 Indirect mechanisms are also likely in the accelerated resolution of allergic lung inflammation, as levels of several inflammatory mediators, including GM-CSF, a pro-survival cytokine for eosinophils, were decreased by LXB4. Of interest, LXA4 has a counter-regulatory effect on GM-CSF signaling in a human eosinophil cell line (EoL-1) and peripheral blood eosinophils.30 LXA4 also inhibits human eosinophils recruitment in vitro31 and murine eosinophils in vivo,10 and can induce natural killer cells to promote human eosinophils apoptosis in asthma to enhance their non-phlogistic clearance from inflamed tissues.32 Together, these findings highlight several cellular mechanisms for lipoxin regulation of allergic airway inflammation.
LXB4 and LXA4 are generated by nasal polyps where a mucosal epithelial 15-lipoxygenase has an important role in their biosynthesis. LXA4 is present in nasal secretions10 and airway samples from human asthmatic patients.9, 33 Eosinophilic donors generate LXA4 preferentially to LXB4,34 however, this is cell type specific as human platelet 12-lipoxygenase converts leukotriene A4 into LXA4 and LXB4 in an equimolar ratio,35 and IL-4 exposed alveolar macrophage 15-lipoxygenase generates LXB4 preferentially to LXA4.36 The nanogram doses of LXB4 used here are within physiological concentrations generated during inflammatory responses and were very potent (∼5 μg kg−1) in comparison with a similar dose of dexamethasone. LXA4 interacts with several receptors, including the G-protein coupled receptor ALX/FPR2. While LXB4 also displays pharmacological properties of a G-protein coupled receptor agonist, it binds to a receptor distinct from ALX/FPR2 that has yet to be identified.
In conclusion, LXB4 displayed pro-resolving actions for mucosal allergic responses in the upper and lower airways. LXB4 is generated in the airway and the present results are the first demonstration of an airway protective effect for LXB4 or that any specialized pro-resolving mediator is active in the nasal mucosa. With the development of LXB4 stable analogs and the limited treatment options currently available, these findings suggest a potential new pro-resolving therapeutic strategy for allergic conditions.
METHODS
Materials. LXA4 and LXB4 were from Cayman Chemical (Ann Arbor, MI) and dexamethasone was from MP Biomedicals LLC (Solon, OH). The lipoxins were stored at −80 °C under a blanket of N2 and their integrity was monitored by UV–vis spectrophotometry and HPLC (Supplementary Figure S1).
Allergic upper and lower airway models. The allergic upper airway model was designed to reflect AR and was a modification of a previously published protocol37 (Figure 1). Briefly, BALB/c female mice (Harlan Laboratories, Jerusalem, Israel), aged 7–8 weeks (20–23 g) were sensitized on day 0 and day 7 by intraperitoneal injection of 200 μl chicken ovalbumin (OVA; 1 mg ml−1) (Amresco, OH) and aluminum hydroxide (alum; 50 mg ml−1) (Acros Organics, Morris Plains, NJ) in phosphate-buffered saline (PBS). Starting on day 14, awake mice were challenged twice a day by intranasal (IN) instillation of 20 μl OVA (25 mg ml−1) daily until day 27. For the allergic lower airway model, a previously published protocol with OVA was used to reflect atopic asthma.38 Briefly, FVB male mice aged 5–7 weeks (Charles River Laboratories) were housed in isolation cages under viral antibody-free conditions. Mice were fed a standard diet (Laboratory Rodent Diet 5001, PMI Nutrition International, St. Louis, MO). Mice were sensitized with intraperitoneal injections of OVA (10 μg) and 1 mg alum in 0.2 ml saline on days 0 and 7. Mice were then challenged on days 14–17 with an aerosol of 6% OVA (25 min).
Treatment protocols. Mice were given intravenous (IV) LXB4 (100 or 1,000 ng), vehicle (0.1% ethanol), or dexamethasone (100 ng). To determine pro-resolving actions, treatments were given after the completion of allergen challenge at peak allergic airway inflammation time: days 28, 29, and 30 in the upper airway model (Figure 1a) or days 18, 19, and 20 in the lower airway model (Figure 7II). Calculation of the resolution interval (Ri) (i.e., time interval for the maximum number of cells to decrease by 50%) was then evaluated as in Bannenberg et al.39 To determine anti-inflammatory actions, LXB4 (100 ng) or vehicle was given IV on protocol days 14, 15, 16, and 17, 30 min before OVA aerosol challenge (Figure 7I). Twenty-four hours after the last IV treatment, mice were euthanized for the collection of airway and systemic specimens. Mouse experiments were approved by the Animal Ethics committee, The Hebrew University, Jerusalem, Israel, and the Harvard Medical Area IRB, Boston, MA (Protocol #03618).
Nasal tissue preparation of cell suspensions and histopathology. As in Miyahara et al.,37 extensive dissection was required to obtain the nasal mucosa cell suspensions (see Supplementary Methods in the Online Data Supplement). Total viable cell numbers were enumerated by Trypan blue exclusion. Nasal tissue sections were prepared and stained for hematoxylin and eosin.
Flow cytometry. Single-cell suspensions from nasal mucosa were resuspended in PBS/0.1% bovine serum albumin seeded in a 96-well plate at 106 cells per ml, blocked with 5% goat serum and stained with Phycoerythrin-conjugated anti-CD107a (eBioscience, San Diego, CA) and anti-Siglec-F (BD Biosciences, San Jose, CA) for the detection of total degranulation40 and eosinophils, respectively. For in vitro assessment of BMMC expression of FCɛRI, cells were stained with anti-FCɛRI (eBioscience) and cell viability was determined by propidium iodide staining (Sigma-Aldrich, Rehovot, Israel). Data were analyzed by a FACSCalibur and CellQuest software (Becton Dickinson, San Jose, CA).
LXA4/LXB4 activity on mouse BMMCs and eosinophils. BMMC and BM eosinophils were obtained as previously described.41, 42 BMMC from BALB/c and FVB mice (1 × 106 per ml) were sensitized overnight with Rat Myeloma IgE (1 μg ml−1; Molecular Probes, Invitrogen, Carlsbad, CA). Cells were washed, resuspended in Tyrode’s buffer and pre-incubated in a 96-well plate (2 × 105 per well) with LXB4, LXA4, or vehicle at the indicated concentrations for 30 min at 37 °C. Cells were then activated by the addition of a goat anti-rat IgG (Fab)2′ antibody (10 μg ml−1) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) (30 min, 37 °C) in Tyrode’s buffer. Degranulation was evaluated by a colorimetric enzymatic β-hexosaminidase release assay.43 For TNF-α release, supernatants were collected 18 h following BMMC activation for evaluation of mTNF-α quantified by ELISA (Peprotech Asia, Israel). For chemotaxis, BM eosinophils were similarly pre-treated and then seeded into the upper chamber of a 24 transwell plate (2 × 105 per well per 100 μl) (Corning, Tewksbury, MA) with a 5.0-μm pore size polycarbonate membrane. Cells were incubated (37 °C, 3 h) to allow migration across the membrane in response to medium only or medium with murine eotaxin (R&D Biosystems, Minneapolis, MN) in the lower wells (50 ng ml−1 per 600 μl). Migrated BM eosinophils were assessed by aspirating a fixed volume from the lower wells and counting the cells by flow cytometer (20 s, duplicate determinations).
Cytospin preparation. Nasal mucosa and BALF cells were resuspended in PBS/2% bovine serum albumin, enumerated by a hemacytometer and cytocentrifuged (1,000 r.p.m., 4 min, r.t.) followed by staining with Modified Wright-Giemsa (Sigma-Aldrich). Total cells, MC, and eosinophil numbers were counted.
Evaluation of serum cytokines/chemokines and OVA-specific IgE levels. Blood samples were collected from the inferior vena cava and sera were stored (−80 °C) until measurement of OVA-specific IgE levels by ELISA (Uscn Life Science Inc., Wuhan, China) and select cytokines/chemokines with Quantibody Mouse cytokine array 1 (Ray Biotech, Norcross, GA).
BALF and lung sample preparation and evaluation. Broncho-alveolar lavage was performed with 2 × 1 ml aliquots of PBS with 0.6 mM EDTA. Lungs were fixed at 25 cm H2O in 10% buffered formalin and paraffin embedded for hematoxylin and eosin and Periodic Acid Schiff staining (Sigma-Aldrich, St. Louis, MO). Cell-free BALF (centrifuged at 2,000 g for 10 min) was coded and select cytokines/chemokines measured by bead array (Pierce Biotechnology, Rockford, IL).
RNA isolation and real-time PCR. Lungs were obtained and snap-frozen RNA was extracted with TRIzol (Invitrogen) and reverse transcribed. The cDNA was used as a template for the amplification of selected genes. The difference between the Ct value for the gene of interest and the respective Ct value for the control gene was then calculated (ΔCt). The fold change was calculated as 2−ΔΔCt. Primers were purchased from Invitrogen: mouse IL-13Rα1: right CGTGCAGAGATTTTCGACAG, left AGCTGTTGGTGCTGCTACTG, IL13Rα2: right GGCAAAGAAGTAACAAAAGGAAT, left CGAATGGAGTGAAGAGGAATG, IL-4R: right AAGCACGCAGATCCAAAATC, left GTGGAGCCTGAACTCGCA, GAPDH: right TTGATGGCAACAATCTCCAC, left CGTCCCGTAGACAAAATGGT, and β-actin: right ATGGAGGGGAATACAGCCC, left TTCTTTGCAGCTCCTTCGTT.
Statistical analysis. All parameters were analyzed using the GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Data are expressed as means±s.e.m.’s. Statistical comparisons were performed using one-way ANOVA, followed by the Tukey post test. When only two groups were compared, statistical differences were determined using the unpaired Student’s t-test. A P-value of <0.05 was considered as significant.
References
Fanta, C.H . Asthma. N. Engl. J. Med. 360, 1002–1014 (2009).
Plaut, M . & Valentine, M.D . Clinical practice. Allergic rhinitis. N. Engl. J. Med. 353, 1934–1944 (2005).
KleinJan, A . et al. United airways: circulating Th2 effector cells in an allergic rhinitis model are responsible for promoting lower airways inflammation. Clin. Exp. Allergy 40, 494–504 (2010).
Palm, N.W ., Rosenstein, R.K . & Medzhitov, R . Allergic host defences. Nature 484, 465–472 (2012).
Locksley, R.M . Asthma and allergic inflammation. Cell 140, 777–783 (2010).
Levy, B.D ., Vachier, I . & Serhan, C.N . Resolution of inflammation in asthma. Clin. Chest. Med. 33, 559–570 (2012).
Edenius, C ., Kumlin, M ., Bjork, T ., Anggard, A . & Lindgren, J.A . Lipoxin formation in human nasal polyps and bronchial tissue. FEBS Lett. 272, 25–28 (1990).
Levy, B.D . et al. Agonist-induced lipoxin A4 generation: detection by a novel lipoxin A4-ELISA. Lipids 28, 1047–1053 (1993).
Planaguma, A . et al. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am. J. Respir. Crit. Care Med. 178, 574–582 (2008).
Levy, B.D . et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4). Nat. Med. 8, 1018–1023 (2002).
Lee, T.H . et al. Lipoxin A4 and lipoxin B4 inhibit chemotactic responses of human neutrophils stimulated by leukotriene B4 and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Clin. Sci. 77, 195–203 (1989).
Lu-Kuo, J.M ., Joyal, D.M ., Austen, K.F . & Katz, H.R . gp49B1 inhibits IgE-initiated mast cell activation through both immunoreceptor tyrosine-based inhibitory motifs, recruitment of src homology 2 domain-containing phosphatase-1, and suppression of early and late calcium mobilization. J. Biol. Chem. 274, 5791–5796 (1999).
Fallon, P.G ., Emson, C.L ., Smith, P . & McKenzie, A.N . IL-13 overexpression predisposes to anaphylaxis following antigen sensitization. J. Immunol. 166, 2712–2716 (2001).
Wills-Karp, M . et al. Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261 (1998).
Kuperman, D.A . et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889 (2002).
Zhu, Z . et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Invest. 103, 779–788 (1999).
Gilmour, J . & Lavender, P . Control of IL-4 expression in T helper 1 and 2 cells. Immunology 124, 437–444 (2008).
Kashiwada, M . et al. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc. Natl. Acad. Sci. USA 107, 821–826 (2010).
Kopf, M . et al. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature 362, 245–248 (1993).
Ramalingam, T.R . et al. Unique functions of the type II interleukin 4 receptor identified in mice lacking the interleukin 13 receptor alpha1 chain. Nat. Immunol. 9, 25–33 (2008).
Busse, W.W . et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 364, 1005–1015 (2011).
Wenzel, S . et al. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 368, 2455–2466 (2013).
Maddox, J.F . et al. Lipoxin B4 regulates human monocyte/neutrophil adherence and motility: design of stable lipoxin B4 analogs with increased biologic activity. FASEB J 12, 487–494 (1998).
Gronert, K . et al. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J. Biol. Chem. 280, 15267–15278 (2005).
Shen, J.C ., Chen, B . & Cohen, N.A . Keratinocyte chemoattractant (interleukin-8) regulation of sinonasal cilia function in a murine model. Int. Forum Allergy Rhinol. 2, 75–79 (2012).
Shen, J.C ., Cope, E ., Chen, B ., Leid, J.G . & Cohen, N.A . Regulation of murine sinonasal cilia function by microbial secreted factors. Int. Forum Allergy Rhinol. 2, 104–110 (2012).
Martin, N . et al. Primary human airway epithelial cell-dependent inhibition of human lung mast cell degranulation. PLoS ONE 7, e43545 (2012).
Gewirtz, A.T ., Fokin, V.V ., Petasis, N.A ., Serhan, C.N . & Madara, J.L . LXA4, aspirin-triggered 15-epi-LXA4, and their analogs selectively downregulate PMN azurophilic degranulation. Am. J. Physiol. 276, C988–C994 (1999).
Ariel, A ., Chiang, N ., Arita, M ., Petasis, N.A . & Serhan, C.N . Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J. Immunol. 170, 6266–6272 (2003).
Starosta, V ., Pazdrak, K ., Boldogh, I ., Svider, T . & Kurosky, A . Lipoxin A4 counterregulates GM-CSF signaling in eosinophilic granulocytes. J. Immunol. 181, 8688–8699 (2008).
Soyombo, O ., Spur, B.W . & Lee, T.H . Effects of lipoxin A4 on chemotaxis and degranulation of human eosinophils stimulated by platelet-activating factor and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Allergy 49, 230–234 (1994).
Barnig, C . et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 5, 174ra126 (2013).
Kazani, S . et al. Exhaled breath condensate eicosanoid levels associate with asthma and its severity. J. Allergy Clin. Immunol. 132, 547–553 (2013).
Serhan, C.N ., Hirsch, U ., Palmblad, J . & Samuelsson, B . Formation of lipoxin A by granulocytes from eosinophilic donors. FEBS Lett. 217, 242–246 (1987).
Romano, M . et al. Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem. J. 296, 127–133 (1993).
Levy, B.D . et al. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J. Clin. Invest. 92, 1572–1579 (1993).
Miyahara, S ., Miyahara, N ., Takeda, K ., Joetham, A . & Gelfand, E.W . Physiologic assessment of allergic rhinitis in mice: role of the high-affinity IgE receptor (FcepsilonRI). J. Allergy Clin. Immunol. 116, 1020–1027 (2005).
Levy, B.D . et al. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J. Immunol. 178, 496–502 (2007).
Bannenberg, G.L . et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 174, 4345–4355 (2005).
Grutzkau, A . et al. LAMP-1 and LAMP-2, but not LAMP-3, are reliable markers for activation-induced secretion of human mast cells. Cytometry A 61, 62–68 (2004).
Jensen, B.M ., Beaven, M.A ., Iwaki, S ., Metcalfe, D.D . & Gilfillan, A.M . Concurrent inhibition of kit- and FcepsilonRI-mediated signaling: coordinated suppression of mast cell activation. J. Pharmacol. Exp. Ther. 324, 128–138 (2008).
Dyer, K.D . et al. Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J. Immunol. 181, 4004–4009 (2008).
Blank, U . & Rivera, J . Assays for regulated exocytosis of mast cell granules. Curr Protoc Cell Biol (Bonifacino J.S., Dasso M., Harford J.B., Lippincott-Schawartz J., Yamada K.M., eds) 15.11.1–15.11.18 Wiley & Sons: New York, (2006).
Acknowledgements
We acknowledge Professor Charles N. Serhan for helpful discussions on this project and critical appraisal of the manuscript, as well as Nour Karra and Jennifer Colby for assisting with sample preparations. This research was supported in part by the US National Institutes of Health grants AI068084, HL68669, and P01-GM095467 (B.D.L.) and by the Aimwell Charitable Fund, UK (F.L-S).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Author contributions
L.K. planned and performed experiments, collected and analyzed data, and wrote the manuscript; O.H. planned and performed experiments, collected and analyzed data, and edited the manuscript; R.P. performed experiments, B.D.L. and F.L-S co-designed the study, performed experiments, analyzed data, and wrote the manuscript.
SUPPLEMENTARY MATERIAL is linked to the online version of the paper
Supplementary information
Rights and permissions
About this article

Cite this article
Karra, L., Haworth, O., Priluck, R. et al. Lipoxin B4 promotes the resolution of allergic inflammation in the upper and lower airways of mice. Mucosal Immunol 8, 852–862 (2015). https://doi.org/10.1038/mi.2014.116
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.116
This article is cited by
-
Influence of physico-chemical properties of two lipoxin emulsion-loaded hydrogels on pre-polarized macrophages: a comparative analysis
Drug Delivery and Translational Research (2024)
-
Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2–IRF pathway
Inflammopharmacology (2022)
-
Cathelicidin preserves intestinal barrier function in polymicrobial sepsis
Critical Care (2020)
