
Abstract
Inflammatory bowel disease (IBD) is characterized for dysregulated intestinal inflammation. Conflicting reports have shown that activation of inflammasome could promote or decrease intestinal inflammation in an acute colitis model, whereas the involvement of inflammasome activation in chronic colitis is poorly understood. In this study, we investigated the role of inflammasome activation in the development of chronic intestinal inflammation by utilizing interleukin-10 (IL-10) knockout (KO) mouse as an animal model, which develops chronic colitis resembling human IBD. We demonstrate the causative link between inflammasome activation and the development of chronic intestinal inflammation. Our results show that mature IL-1β protein levels were significantly increased in all colon sections from IL-10-deficient mice compared with that of wild-type mice. We found that inhibition of inflammasome activities with IL-1 receptor antagonist or caspase-1 inhibitors suppressed IL-1β and IL-17 production from inflamed colon explants. Furthermore, blocking inflammasome activation with caspase-1 inhibitor in vivo significantly ameliorated the spontaneous colitis in IL-10 KO mice. Taken together, these observations demonstrate that inflammasome activation promotes the development of chronic intestinal inflammation.
Similar content being viewed by others


Introduction
Inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), is a chronic disease that results from dysregulated immune response to commensal microflora.1, 2, 3, 4, 5 It has been estimated that 1 to 2 million individuals in North America suffer from IBD, with the peak of onset occurring between 15 and 30 years of age. Furthermore, the number of individuals diagnosed with CD has been increasing steadily during the past several decades. Although the etiology for IBD is unknown, it is believed that genetic and environmental factors as well as the immune system contribute to the development of intestinal inflammation.
Genetic studies have identified many loci in the genome associated with intestinal inflammation. Most of IBD-associated genes are involved in inflammation and immunity, which include interleukin-10 (IL-10), NOD-like receptors (NLRs), Toll-like receptors (TLRs), IL-1 receptor (IL-1R), and IL-23. In addition, pathways related to stress response and autophagy, such as XBP1 and ATG16L1, are involved in IBD pathogenesis.1, 5, 6, 7 Recent studies have demonstrated that IL-17, produced by T helper type 17 (Th17) cells and innate lymphoid cells, contributes to the pathogenesis of intestinal inflammation. In contrast, IL-10, produced by regulatory T cells and innate immune cells, has a critical role in controlling intestinal inflammation and maintaining intestinal homeostasis, inasmuch IL-10-deficient mice spontaneously develop colitis, resembling the pathogenesis of human CD.8, 9, 10
The members of the cytosolic NLR family have been identified as the critical regulators of inflammation and cytokine production. Remarkably, the NLR protein CARD15/NOD2 has been identified to be associated with CD.11, 12 Recently, mutation in NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) has been reported to be associated with CD.13 In addition, caspase-1, which regulates the secretion of biologically active IL-1β and IL-18, has been identified as a central mediator in dextran sodium sulfate (DSS)-induced colitis.14 The proinflammatory cytokines such as IL-1β has a critical role in the pathogenesis of IBD. A number of clinical studies show increased IL-1β secretion from colonic tissues and macrophages of IBD patients. The IL-1β levels are also correlated with the diseases severity of IBD.9, 15, 16, 17, 18 Furthermore, the clinical efficacy with therapies targeting IL-1β has demonstrated a critical role of IL-1 in certain autoimmune diseases. Because of its potential in inducing proinflammatory response and to cause damage to the host, IL-1β production is tightly controlled at multiple levels. Production of mature IL-1β requires at least two signals: the first signal is initiated by TLR ligands or endogenous molecules that induce pro-IL-1β gene expression; the second signal includes very diverse stimuli that activate inflammasome, leading to IL-1β maturation.19, 20, 21 Inflammasome is a multimolecular complex, composed of an NLR protein, the adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase-1, which controls processing of proinflammatory cytokine IL-1β and IL-18. In addition to NLRP3, other NLRs such as NLRC4 and NLRP6 can form inflammasome with ASC and caspase-1 in response to different stimuli.20, 22 While functioning as an intracellular defense system against microbe infection, inflammasomes have a critical role in complex diseases including autoimmune diseases, metabolic syndrome, and cancer.23, 24, 25
However, the role of inflammasomes in acute intestinal inflammation remains controversial, and the involvement of inflammasomes and inflammasome-mediated IL-1β in chronic colitis is poorly understood. Although several studies show that mice deficient for inflammasome components including NLRP3, ASC, and caspase-1 are highly susceptible to acute colitis induced by DSS, one study demonstrates the opposite results showing that defects in NLRP3 inflammasome protect mice from DSS-induced acute colitis.23, 26, 27, 28 On the other hand, clinical studies show increased levels of IL-1β in inflamed mucosal tissues from IBD patients compared with normal tissues.9, 15, 16, 17, 18 In this study, we examined the role of inflammasome activation in the development of chronic colitis. Our results show that inflammasome activation promotes the development of chronic intestinal inflammation in IL-10 knockout (KO) mice. We further demonstrated that inhibition of inflammasome activities with IL-1R antagonist (IL-1Ra) or caspase-1 inhibitors suppressed intestinal inflammation.
Results
Spontaneous colitis is associated with increased inflammasome activation and IL-1β production
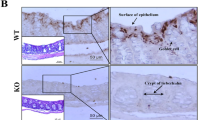
To investigate the role of inflammasome activation in the development of chronic colitis, we utilized IL-10 KO mouse (B6.129P2-Il10tm1Cgn/J) as an animal model of chronic intestinal inflammation. IL-10 KO mice spontaneously develop chronic intestinal inflammation under specific pathogen-free conditions in our animal facility. The phenotypes of chronic colitis were more evident when IL-10 KO mice were 12 weeks or older. IL-10-deficient mice generally displayed the phenotype of significant weight loss and diarrhea. Some animals also showed rectal prolapse and severe sign of illness. As shown in Figure 1a, hematoxylin and eosin (H&E) staining showed that IL-10 KO mice at the age of 16 weeks developed intestinal inflammation in both the small intestine and the colon tissues. Accordingly, IL-10 KO mice displayed significant weight loss (Figure 1b) and markedly increased pathology scores in the colon, mesenteric lymph nodes (MLNs), and the small intestine (Figure 1c).

Spontaneous colitis is associated with increased levels of interleukin-1β (IL-1β) protein in the intestinal system. (a) Hematoxylin and eosin (H&E) staining of colon and small intestine sections from 16-week-old wild-type (WT) and IL-10 knockout (KO) mice. The marked colitis (mucosal ulceration, loss of villi, and decreased crypts) was observed in IL-10 KO mice. (b) Body weight and (c) pathologic inflammation scores in the colon, small intestine, and mesenteric lymph node (MLN) from WT and IL-10 KO at the age of 16 weeks. (d) Immunostaining of IL-1β in colon and small intestine sections from 16-week-old WT and IL-10 KO mice. (e) Colon tissues from WT and IL-10 KO mice at the age of 16 weeks were homogenized, and analyzed by western blot analysis for procaspase-1 and mature caspase-1 as well as pro-IL-1β and mature IL-1β proteins. Levels of IL-1β protein in homogenates of (f) proximal, (g) middle, and (h) distal colon, as well as small intestinal (i) from 16-week-old WT and IL-10 KO mice were analyzed by enzyme-linked immunosorbent assay (ELISA). Data represent mean of the pool of two independent experiments (n=12 for WT and IL-10 KO mice), and results for individual mouse are illustrated. Statistical significance is indicated as *P<0.05, **P<0.01 (Student’s t-test). Bar=100 μm.
Next, we performed immune staining to determine if intestinal tissues with chronic colitis have increased levels of IL-1β. Immunohistochemical analysis revealed a significant increase in IL-1β staining in colon and small intestinal sections from IL-10 KO mice, as compared with that from wild-type (WT) mice (Figure 1d). Interestingly, our staining results show that both innate immune cells in the lamina propria and submucosal sections and epithelial cells were potential sources of IL-1β production. IL-1β is first generated as cytosolic precursors that require cleavage by the protease caspase-1 to become the biologically active cytokine.19, 20, 21 To distinguish between mature and premature IL-1β in colon tissues, we performed western blot analysis on colon tissues from WT and IL-10 KO mice. We found that there were significantly more mature IL-1β and caspase-1 proteins in homogenized colon tissues of IL-10 KO mice (Figure 1e).
To further determine that chronic colitis is associated with increased production of IL-1β protein, small and large intestinal tissues were homogenized and assayed by IL-1β-specific enzyme-linked immunosorbent assay (ELISA). We found that IL-1β protein levels were significantly increased in all colon sections including proximal, middle, and distal colons from IL-10 KO mice. Particularly, small intestinal tissues from IL-10 KO mice also secreted significantly high levels of IL-1β protein (Figure 1f–i). In contrast, intestinal tissues from WT mice had very low or below detection levels of IL-1β. Collectively, these data suggest that inflammasome activity and IL-1β production were enhanced in colitic intestinal tissues of IL-10 KO mice, prompting us to examine closely the roles of IL-10 in modulating the activation of inflammasomes.
IL-10 inhibits inflammasome activation and IL-1β production
Because of the high levels of IL-1β protein in colitic tissues of IL-10 KO mice, we hypothesized that enhanced or prolonged inflammasome activation contributes to the development of chronic colitis in IL-10 KO mice. To test this hypothesis, we first examined inflammasome activation and IL-1β processing in IL-10-deficient macrophages in vitro. Although several types of cells including macrophages, dendritic cells, and epithelial cells are potential sources of IL-1β, this study focused on the inflammasome activation and IL-1β production in macrophages because intestinal inflammation is associated with infiltration of myeloid cells, including macrophages, which produce a variety of inflammatory cytokines in response to intestinal microbes. Another reason is that inflammasome was initially identified in macrophages, and this type of cells can have robust inflammasome activation and IL-1β production.29, 30 Bone marrow-derived macrophages (BMDMs) from WT or IL-10 KO mice were, respectively, primed with lipopolysaccharide (LPS) to induce the expression of pro-IL-1β, and then stimulated with ATP for 1 h to activate NLRP3 inflammasome. As shown in Figure 2a, LPS/ATP-induced secretion of IL-1β was significantly increased in IL-10-deficient BMDMs. To determine if increased IL-1β production was because of enhanced activation of inflammasome, we examined caspase-1 and IL-1β processing in WT and IL-10-deficient macrophages. When supernatants from the BMDM culture were analyzed by western blot, we found that IL-10-deficient BMDMs released significantly more mature IL-1β and caspase-1 upon LPS/ATP stimulation than WT cells (Figure 2b). This result indicates that IL-10 can regulate IL-1β production through modulation of inflammasome activities.

IL-10 deficiency leads to increased NLRP3 (NACHT, LRR, and PYD domains-containing protein 3) inflammasome activation and interleukin-1β (IL-1β) production. (a) Wild-type (WT) and IL-10-deficient bone marrow-derived macrophages (BMDMs) were primed with lipopolysaccharide (LPS) for 4 h, and then were treated with ATP for 1 h to induce inflammasome activation. The concentration of IL-1β in culture supernatants was assayed by enzyme-linked immunosorbent assay (ELISA). (b) WT and IL-10-deficient BMDMs were stimulated with LPS and ATP as in panel a. Inflammasome activation was determined by western blot analysis of activated caspase-1 and processed IL-1β in the culture supernatants of BMDMs stimulated with LPS plus ATP. (c) Caspase-1 mRNA levels in BMDMs from WT and IL-10 KO mice treated as described in panel a were analyzed by reverse transcription-polymerase chain reaction (RT-PCR). (d and e) WT and IL-10-deficient BMDMs were primed with LPS for 4 h, and then cells were treated with Alum or monosodium urate (MSU) crystal for 4 h. The concentration of IL-1β in culture supernatants was assayed by ELISA. Representative data from one of at least three independent experiments are shown. **P<0.01 (Student’s t-test).
We also noticed that procaspase-1 protein levels were significantly increased in cells from IL-10 KO mice. To determine whether caspase-1 gene expression and activation are affected by IL-10, we analyzed caspase-1 mRNA expression in BMDMs by quantitative real-time polymerase chain reaction. As shown in Figure 2c, the caspase-1 mRNA expression level was significantly increased in IL-10-deficient macrophages activated by LPS and ATP, indicating the ability of IL-10 to inhibit caspase-1 gene expression in activated macrophages. Taken together, our results suggest that IL-10 can regulate IL-1β production through multiple mechanisms.
In addition to ATP, NLRP3 can be activated by a number of different stimuli. We found that defects in IL-10 production also led to increased IL-1β production in macrophages treated with LPS plus Alum or monosodium urate crystal (Figure 2d and e). These results demonstrate that IL-10 functions as a negative regulator of NLRP3 inflammasome activity triggered by different stimuli. Next, we examined effects of exogenous IL-10 on NLRP3 inflammasome activation. Recombinant IL-10 was added at different time points during or before LPS and ATP stimulation. Our data show that adding just 1 h before ATP treatment, IL-10 was able to inhibit IL-1β processing (Figure 3a and b), indicating that early signaling events induced by IL-10 suppressed inflammasome activation directly. Similarly, we found that IL-10 inhibited LPS and Alum-induced IL-1β production (Figure 3c). As an alternative approach to examine the direct effect of IL-10 on inflammasome activation, we performed an inflammasome-reconstitution assay in 293T cells. In this system, 293T cells were transfected with NLRP3/ASC/caspase-1 and IL-1β plasmids, and thus any effects of a molecule on caspase-1 and IL-1β processing could be tested. As shown in Figure 3d, pre-treatment with IL-10 for 1–8 h significantly inhibited IL-1β production in our reconstitution system. Taken together, these results suggest that IL-10 directly downregulates inflammasome activity.

IL-10 inhibits inflammasome activation and interleukin-1β (IL-1β) production. (a and b) Wild-type (WT) bone marrow-derived macrophages (BMDMs) were primed with lipopolysaccharide (LPS) for 4 h, and then treated with ATP for inflammasome induction. One hour before adding ATP, cells were treated with 25 ng ml−1 of recombinant murine IL-10. The concentration of IL-1β in culture supernatants was assayed by enzyme-linked immunosorbent assay (ELISA). Caspase-1 and IL-1β processing and releasing were analyzed by western blot analysis. (c) WT BMDMs were primed with LPS for 4 h, and then treated with Alum for 4 h. One hour before adding Alum, cells were treated with 25 ng ml−1 of recombinant murine IL-10. The concentration of IL-1β in culture supernatants was assayed by ELISA. (d) The 293T cells were reconstituted with inflammasome components (NACHT, LRR and PYD domains-containing protein 3 (NLRP3), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), caspase-1, and pro-IL-1β plasmids). Thirty-six hours after transfection, cells were pretreated with IL-10 for different time points as indicated, and then with ATP for 1 h. The IL-1β level in the supernatants was determined by ELISA. Results are reported as mean±s.d. of triplicate samples from one representative experiment of three independent experiments. Statistical significance is indicated as *P<0.05; **P<0.01 (Student’s t-test).
Inflammasome-mediated IL-1β promotes IL-17 production in IL-10 KO mice
Recent studies indicate that IL-17, produced either by Th17 cells or innate lymphoid cells, contributes to the pathogenesis of chronic colitis in animal models and in human IBD patients.31, 32, 33 Although IL-1 has been shown to promote the differentiation of naive T cells into Th17 cells,9, 34 results from other labs and ours have demonstrated that IL-10 produced from innate immune cells and T cells can limit IL-17 production from Th17 cells.35, 36 As our results mentioned above indicate that chronic colitis is associated with enhanced inflammasome activation and IL-1β production, we investigated whether inflammasome-derived IL-1β modulates IL-17 production during intestinal inflammation. We observed that IL-10 KO mice have significantly enlarged MLNs, which contain increased number of immune cells as compared with WT mice. As intestinal tissues from IL-10-deficient mice have increased inflammasome activity, we reasoned that LPS stimulation could further boost pro-IL-1β levels, consequently IL-1β production, which may induce more IL-17 production. Therefore, when stimulated with anti-CD3 ex vivo, IL-10-deficient MLN cells are capable of producing more IL-17. In contrast, most WT MLN T cells are under resting state or naive T cells, which may need costimulation of CD3 and CD28 as well as cytokines (tumor growth factor-β and IL-6) to induce the production of IL-17. When MLN cells from IL-10-deficient mice were treated with both anti-CD3 antibody and LPS, IL-17 production was strongly enhanced (Figure 4a). To further prove that inflammasome-derived IL-1β promotes IL-17 production, we used IL-1β in combination with anti-CD3 antibody to stimulate MLN cells; Figure 4b shows that IL-1β induced significant amount of IL-17 from anti-CD3-stimulated IL-10-deficient MLN cells. In contrast, WT MLN cells produced much less IL-17.

Inhibition of inflammasome activities leads to reduced interleukin-17 (IL-17) production from mesenteric lymph node (MLN) cells of IL-10 knockout (KO) mice. (a and b) MLN cells from wild-type (WT) and IL-10 KO mice were stimulated with anti-CD3 antibody in combination with lipopolysaccharide (LPS) (100 ng ml−1) or recombinant IL-1β (25 ng ml−1) as indicated for 48 h. The production of IL-17 was measured by enzyme-linked immunosorbent assay ELISA. (c and d) MLN cells from IL-10-deficient mice were treated with anti-CD3 antibody or LPS in the presence of IL-1R antagonist (IL-1Ra) or caspase-1 inhibitor Z-VAD-FMK (ZVAD), and IL-17 production was measured 48 h after treatment. Results are reported as mean±s.d. of the pool of two experiments (5–6 mice per group). **P<0.01 (Student’s t-test).
Our results suggest that increased inflammasome activation and IL-1β production contribute to increased IL-17 production in chronic colitis. We therefore tested if blocking inflammasome activation and IL-1β pathways inhibits IL-17 production and intestinal inflammation. We utilized two independently blocking agents to evaluate the consequences of suppressing IL-1β production and signaling. These agents included IL-1Ra, which inhibits IL-1β activity, and a caspase inhibitor Z-VAD-FMK, which inhibits IL-1β processing. As shown in Figure 4c and d, treatment with IL-1Ra or Z-VAD-FMK each led to reduced IL-17 production from IL-10-deficient MLN cells stimulated with LPS and anti-CD3 antibody ex vivo. These results indicate that IL-10 and inflammasome-processed IL-1β reciprocally regulate IL-17 production.
Blocking inflammasome activation and IL-1β pathways inhibit IL-17 production in intestinal tissues of chronic colitis
The effects of caspase inhibitors on MLN cells promoted us to determine whether blocking inflammasome activation inhibits IL-1β and IL-17 production from inflamed intestinal tissues. To address this question, we evaluated the effects of IL-1Ra on IL-1β and IL-17 production from colon explants. As shown in Figure 5a and b, when organ explants of colon and small intestine were cultured for 24 h in the medium without any treatment, colon and small intestinal tissues from IL-10 KO mice secreted substantial amount of IL-1β compared with that from WT mice. Owing to increased inflammasome activation in intestinal organs of IL-10-deficient mice, we found that adding LPS to organ explants significantly enhanced IL-1β production. Furthermore, our results show that IL-1Ra inhibited LPS-induced IL-1β production from organ explants. Those results indicate that inflammasome-induced IL-1β may form a positive feedback forward loop to induce more IL-1β production in intestinal tissues.

Blocking interleukin-1 receptor (IL-1R) function inhibits IL-1β and IL-17 production from inflamed intestinal tissues of IL-10 knockout (KO) mice. (a and b) Colon and small intestine isolated from wild-type (WT) and IL-10 KO mice were stimulated with lipopolysaccharide (LPS) in the presence or absence of IL-1R antagonist (IL-1Ra). After 24 h, IL-1β production was determined by enzyme-linked im+munosorbent assay (ELISA). (c and d) Colon and small intestinal explants were treated with LPS or IL-1Ra, and IL-17 production was measured. Results are reported as mean±s.d. of the pool of two experiments (5–6 mice per group). *P<0.05; **P<0.01 (Student’s t-test).
Next, we determined if blocking inflammasome-mediated IL-1β release could suppress IL-17 production from intestinal tissues. Colon and small intestinal explants from WT and IL-10 KO mice were treated with IL-1Ra, and assayed for the concentration of IL-17 by ELISA. We found that colon tissues with chronic inflammation from IL-10 KO mice produced more IL-17. Furthermore, LPS treatment increased IL-17 production from colon explants. Notably, when the function of inflammasome-derived IL-1β was blocked with IL-1Ra, LPS-induced IL-17 secretion was significantly suppressed (Figure 5c and d). Taken together, our data suggest that inflammasome-derived IL-1β promotes IL-17 production from intestinal tissues.
Inhibition of inflammasome activation decreases chronic colitis
To test our hypothesis that blocking inflammasome activation could suppress the spontaneous colitis in vivo, we treated IL-10 KO mice at the age of 16 weeks with Ac-YVAD-cmk (acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone), a caspase-1-specific inhibitor. At week 18, intestinal tissues from treated or untreated mice were examined histologically for signs of inflammation. As shown in Figure 6a, the colon from untreated IL-10 KO mice was edematous, thickened, and shortened by inflammation. In contrast, inflammasome blockade significantly ameliorated the spontaneous colitis. Histological examination revealed infiltration of inflammatory cells into the mucosa and submucosa in the colon of saline-treated IL-10 KO mice, while the Ac-YVAD-cmk-treated mice showed much less inflammation in the colon tissues, and significantly improved pathology scores. (Figure 6b and c). Immunohistochemistry staining also demonstrated decreased IL-1β protein expression in the colons of the Ac-YVAD-cmk-treated mice (Figure 6d). Moreover, Ac-YVAD-cmk treatment significantly decreased IL-17 production from IL-10-deficient MLN cells (Figure 6e). These results indicate that inhibition of inflammasome activation decreases the spontaneous colitis in IL-10 KO mice.

Inflammasome blockade ameliorates the spontaneous colitis in interleukin-10 (IL-10)-deficient mice. (a) IL-10 knockout (KO) mice at 16 weeks old were injected intraperitoneally with caspase-1-specific inhibitor Ac-YVAD-cmk (acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone) for 2 weeks. Macroscopic colon pathology and colon length from IL-10 KO mice treated with or without caspase-1 inhibitor were determined. (b) Hematoxylin and eosin (H&E)-stained sections and (c) pathology scores of colon and small intestine sections from 16-week-old IL-10 KO mice or IL-10 KO mice treated with Ac-YVAD-cmk for 2 weeks. (d) IL-1β immunostaining of colon and small intestine sections from 16-week-old IL-10 KO mice treated with caspase-1 inhibitor as in panel a. (e) Mesenteric lymph node (MLN) cells from IL-10 KO mice treated with or without caspase-1 inhibitor Ac-YVAD-cmk were stimulated with anti-CD3 antibody for 48 h. The production of IL-17 was measured by enzyme-linked immunosorbent assay (ELISA). Data represent mean of the pool of two independent experiments (n=5–6 for treated or untreated IL-10 KO mice). Statistical significance is indicated as *P<0.05 (Student’s t-test). Bar=100 μm.
IL-1β-induced Th17 cells contribute to colitis in IL-10 KO mice
To further confirm that IL-1β-induced pathogenic T cells contribute to the development of chronic colitis in IL-10 KO mice, we would like to determine whether transferring of IL-17-producing cells from IL-10 KO mice into caspase-1 inhibitor-treated IL-10 KO mice would increase the colitis severity. In transfer experiments, we utilized MLN cells, which contain IL-17-producing T cells, and possibly other colitic cells, from untreated IL-10 KO mice. IL-10 KO mice at the age of 16 weeks were pretreated with caspase-1-specific inhibitor Ac-YVAD-cmk for 2 weeks, which led to ameliorated colitis. Then, Ac-YVAD-cmk-treated IL-10 KO mice were injected intraperitoneally with MLN cells from untreated IL-10 KO mice. The receipt mice were examined for clinical signs of colitis again. At week 20, mice were killed, and intestinal tissues and MLN from transferred and untransferred mice were examined histologically for intestinal inflammation. In this reciprocal set of experiments, transfer of pathogenic MLN cells led to a significant enhancement of colitis in mice pretreated with the caspase-1-specific inhibitor as indicated by increased intestinal pathology macroscopically, and more severe pathology scores (Figure 7a–c). H&E staining revealed that Ac-YVAD-cmk-treated IL-10 KO mice receiving pathogenic MLN cells had similar inflammatory symptom in the colon and small intestine with IL-10 KO mice, while the Ac-YVAD-cmk-treated mice showed much less inflammation in the intestinal tissues (Figure 7c and d). Furthermore, immunohistochemistry staining also demonstrated increased IL-1β protein expression in the colons and small intestines of the Ac-YVAD-cmk-treated mice transferred with pathogenic MLN cells (Figure 7e). These results indicate that inflammasome/IL-1β-induced T cells contribute to colitis in IL-10 KO mice.

Interleukin1β (IL-1β)-induced IL-17 production enhanced colitis in IL-10 knockout (KO) mice. (a and b) Macroscopic colon pathology and colon length from Ac-YVAD-cmk (acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone)-treated IL-10 KO mice transferred with mesenteric lymph node (MLN) cells from IL-10 KO mice. (c) Pathologic inflammation scores in the colon, small intestine, and MLN from Ac-YVAD-cmk-injected IL-10 KO mice treated with or without IL-10-deficient MLN cells. (d) Hematoxylin and eosin (H&E) staining and (e) IL-1β immunostaining of colon and small intestine sections from Ac-YVAD-cmk-treated IL-10 KO mice transferred with or without MLN cells of IL-10 KO mice. Data represent mean of the pool of two independent experiments (n=5 for MLN cells treated or untreated with Ac-YVAD-cmk-injected IL-10 KO mice). Statistical significance is indicated as *P<0.05 (Student’s t-test).
Discussion
In this report, we have demonstrated that chronic colitis is associated with enhanced inflammasome activation and IL-1β production. We further demonstrate the direct inhibitory role of IL-10 in inflammasome activation in macrophages and in intestinal tissues. Our results indicate that without the inhibitory molecule IL-10, inflammasomes in the intestinal tissues undergo prolonged activation, because of constant stimulation from microbes, leading to the chronic colitis. Finally, our results show that caspase-1-specific inhibitor could suppress the chronic colitis in IL-10 KO mice.
The pathogenic roles of NLRP3 inflammasome we described in the IL-10 KO mice are different from protective roles for inflammasome activation in acute intestinal inflammation of DSS-treated mice reported recently. Several studies reported that mice deficient for inflammasome components including NLRP3 exhibit enhanced DSS-induced colitis compared with WT mice.22, 26, 27, 28, 37 However, one study suggests that DSS-induced intestinal inflammation is mediated by NLRP3 inflammasome. Bauer et al.23 reported that NLRP3 KO mice treated with DSS had attenuated colitis and lower mortality. The reasons for this discrepancy are unclear, but it may result from variations in mouse genetic backgrounds and intestinal microflora. Importantly, our results are consistent with several studies using chronic colitis models. For instance, an early study by Siegmund et al.14 demonstrated that IL-1-converting enzyme (caspase-1) deficiency led to protection from acute and chronic DSS-induced colitis. They found that IL-1-converting enzyme/caspase-1 KO mice displayed a more than 50% decrease in the clinical scores as assessed by weight loss, diarrhea, rectal bleeding, and colon length. In another study, Vijay-Kumar et al.38 and Carvalho et al.39 suggested that IL-1β promoted susceptibility of TLR5-deficient mice to colitis. The susceptibility of TLR5 KO mice to colitis depends on gut microbiota. Interestingly, Carvalho et al.39 found that administration of anti-IL-10 receptor blocking antibody led to severe uniform intestinal inflammation in both colitis-susceptible and -resistant strains of TLR5 KO mice. The authors further showed that IL-1β was crucial for this colitis model, because IL-1R and TLR5 double KO mice were completely protected from colitis occurred in TLR5 KO mice upon IL-10 signaling blockade. The contradicting observations imply that inflammasomes may have colitis-promoting or -suppressing roles in different conditions.
The difference between these results may result from choice of colitis models and variation in commensal microflora. In addition, the mechanisms responsible for DSS-induced acute colitis and IL-10 deficiency-mediated chronic colitis may be different. In acute colitis model, DSS in drinking water causes direct damage to the epithelial barrier, which allows the microflora to stimulate innate cells, leading to infiltration of myeloid cells and massive inflammation. During this chemically induced inflammation, the IL-1β and IL-18 produced by macrophages and dendritic cells in the intestinal tissues are essential for tissue repair and the homeostasis of epithelial barrier.40 Therefore, inflammasomes protect the intestinal system from tissue injury caused by pathogens. In this context, mice deficient for inflammasome components such as caspase-1 and NLRP3 develop more severe colitis in response to acute damage mediated by DSS. In contrast, in chronic colitis, as occurred in IL-10 KO mice, inflammasome and IL-1β may induce prolonged intestinal inflammation and the development of pathogenic Th17 cells. We found that intestinal tissues with chronic inflammation from IL-10 KO mice produced more IL-17. Furthermore, when inflammasome activation was blocked with caspase-1-specific inhibitors, or the function of inflammasome-derived IL-1β was blocked with IL-1Ra, IL-17 production from intestinal tissues was significantly reduced. Our data suggest that inflammasome-derived IL-1β promotes IL-17 production in intestinal tissues and the development of colitis.
On the basis of studies by other and our groups, we propose that IL-10 may influence IL-1β production through both gene transcription and direct regulating inflammasome activation. IL-10 has been shown as an important anti-inflammatory cytokine in maintaining homeostasis of immune system. Our previous studies have shown that IL-10 inhibits the expression of inflammatory cytokines including pro-IL-1β induced by LPS.41 Interestingly, type I interferon, which has been shown to inhibit inflammasome activation via IL-10-dependent or -independent mechanisms,42, 43 can also limit intestinal inflammation.44, 45 In this study, we found that transcriptional induction of the caspase-1 gene was also inhibited by IL-10. These results suggest that one of the mechanisms by which IL-10 regulates inflammasome activities and IL-1β production is by suppressing gene expression of inflammasome components. Importantly, results from this study also indicate that IL-10 can directly inhibit NLRP3 inflammasome activation and IL-1β processing, suggesting that IL-10 signaling molecules interact directly or indirectly with inflammasome pathways. At present, the molecular mechanisms of IL-10-mediated inhibition of inflammasomes remain unknown. We think our in vivo and in vitro systems described in this study will help to unravel the mechanisms responsible for negative regulation of inflammasome activation by IL-10.
Although this study focused on the role of NLRP3 inflammasome and IL-1β in the chronic colitis in IL-10 KO mice, other inflammasomes such as NLRC4 or inflammasome-processed cytokine IL-18 may affect intestinal inflammation as well. In addition to innate immune cells, intestinal epithelial cells have been shown to express inflammasome components and have inflammasome activation in response to intestinal microbes.22, 37, 46 Given the contrasting data regarding the role of inflammasomes in intestinal inflammation and homeostasis, cell type-specific mutations may shed light on the function of inflammasome in epithelial cells and innate cells. Emerging evidence has demonstrated that the commensal microflora has an important role in the broad aspects of immunity including the intestinal immune system.1, 21, 47 The apparent contradiction about the role of inflammasomes in mucosal immunity may be heavily influenced by gut microflora, although further studies are needed to resolve this issue. As IBD is a heterogeneous disease, it is possible that in some IBD patients, genetic mutations or environmental factors may cause reduced inflammasome activation and IL-1β production, which compromise the integrity of the epithelial cell barrier against microflora, leading to intestinal inflammation. Whereas in other IBD patients, genetic factors and commensal microflora may cause prolonged or unregulated inflammasome activation and increased IL-1β production, generating uncontrolled inflammation in the intestine system.
Methods
Reagents. All chemicals used in the present study were purchased from Sigma (St Louis, MO), unless otherwise noted. Ultrapure LPS, Pm3C, ATP, monosodium urate crystal, caspase inhibitors Z-VAD-FMK, Ac-YVAD-cmk, and puno-NLRP3 were purchased from Invivogen (San Diego, CA). Murine or human IL-1β and IL-10 was from PeproTech (Rocky Hill, NJ); anti-CD3 antibody was from BD Biosciences (San Diego, CA); human or murine IL-1β ELISA Kits were from R&D Systems (Minneapolis, MN) or eBioscience (San Diego); and Alum (Imject Alum) was purchased from Pierce Biochemicals (Rockford, IL). Antibodies to human IL-1β, human caspase-1, mouse IL-1β, and mouse caspase-1 were from R&D Systems or Santa Cruz Biotechnology (Santa Cruz, CA).
Mouse. Both WT C57BL/6 and IL-10 KO (B6.129P2-Il10tm1Cgn/J) mice were purchased from the Jackson Laboratories (Bar Harbor, ME), and bred at our Hollings Cancer Center animal facility for the past 2 years. All mice were maintained at Medical University of South Carolina Hollings animal facility under specific pathogen-free conditions. In all experiments, IL-10 KO mice and WT mice are kept at the same rack of an animal room, but not in the same cage. IL-10 KO mice spontaneously develop a chronic IBD under specific pathogen-free conditions in our animal facility. The phenotypes of chronic enterocolitis were more evident when IL-10 KO mice were 12 weeks or older. Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Medical University of South Carolina (Charleston, SC), and were conducted in accordance with federal regulations as well as institutional guidelines and regulations on animal studies.
For caspase-1 inhibition, IL-10 KO mice at the age of 16 weeks were injected intraperitoneally with caspase-1 inhibitor Ac-YVAD-cmk at 1.25 mg kg−1 for 2 weeks. For MLN cell transfer experiments,48 cells were isolated from the MLNs of IL-10 KO mice at the age of 16 weeks, and washed. A single-cell suspension of 5 × 106 MLN cells were injected intraperitoneally into IL-10 KO mice pretreated with a caspase-1 inhibitor. Mice will be monitored for clinical signal of colitis. Two weeks after cell transfer, mice were killed and intestinal inflammation was analyzed.
BMDMs. BMDMs were differentiated as described previously.41, 49 Briefly, murine bone marrow cells were cultured for 7 days in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, penicillin, streptomycin, and 20% conditioned media from L929 cells overexpressing macrophage colony-stimulating factor. To induce IL-1β processing and production, differentiated BMDMs were primed with 100–250 ng ml−1 ultrapure LPS for the 4–6 h, and then cells were stimulated with ATP (1 mM) for 1 h, or monosodium urate crystals (200 μg ml−1) and Alum (40 μg ml−1) for 4 h.
Intestinal tissues and culture. Colons and small intestines were dissected from mice and flushed with cold phosphate-buffered saline containing penicillin/streptomycin to remove fecal contents, and then opened lengthwise and washed extensively. Equivalent amounts of tissue were distributed into wells of a 24-well tissue culture plate in RPMI-1640 supplemented with 10% fetal calf serum, penicillin (100 U ml−1), and streptomycin (100 U ml−1). Organ explants were incubated at 37 °C for 24 or 48 h. Supernatants were collected and assayed for cytokine levels by ELISA, and concentrations were normalized to the weight of the explants.
For tissue homogenates, small intestinal or colonic segments were placed in phosphate-buffered saline with a protease inhibitor cocktail, and minced with dissection scissors. Tissues were then homogenized by a tissue homogenizer, and centrifuged at 13,000 r.p.m. for 15 min. The levels of IL-1β in tissue homogenates were determined by ELISA, and concentrations were normalized to the weight of the tissues.
MLN cells were cultured in the presence of plate-bound anti-CD3ε antibody for 48 h. In some experiments, MLN cells were treated with LPS, IL-1Ra, or caspase inhibitor Z-VAD-FMK. IL-1β and IL-17A levels in the culture supernatants were measured by ELISA.
Inflammasome reconstitution in 293T cells. Briefly, 293T cells were transfected with expression plasmids for human NLRP3 (Invivogen), ASC (Origene, Rockville, MD), caspase-1 (Addgene, Cambridge, MA), and IL-1β. At 24 h after transfection, cells were stimulated with ATP for 1 h, then supernatants were assayed for IL-1β production, and finally cell lysates were analyzed by immunoblot with antibody to IL-1β and caspase-1.
Histology and immunohistochemistry. Colons and small intestines were dissected from mice and flushed with cold phosphate-buffered saline to remove fecal contents, and quickly frozen. Sections were stained with H&E for routine histological analysis, and were immunostained with anti-IL-1β for IL-1β protein levels in intestinal tissues. Positive immunostaining was detected with a streptavidin–biotin immunoperoxidase system (Vector Laboratories, Burlingame, CA) according to the manufacturer’s protocols. For histopathologic analysis, gut pathology score is defined by summation of two parameter scores to achieve a range of 0–6: inflammation (0, no inflammation; 1, neutrophil rarely discernible; 2, presence of neutrophils in every high power field; 3, collection of >10 neutrophils in any given area) and tissue destruction (0, normal structure; 1, villus length shortened by 50%; 2, flattened mucosa with loss of villi; 3, evidence of submucosa ulceration).50
Western blot and ELISA. Cell lysates, cell culture supernatants, or intestinal homogenates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot with specific antibodies as described.41 Cytokine concentration in culture supernatants was determined by cytokine specific ELISA Kit (eBioscience or R&D Systems) per the manufacturer’s instructions.
Statistical analysis. All data were analyzed via the Student’s t-test to determine differences among experimental groups. A treatment difference was considered significant when the P-value was <0.05.
References
Jostins, L. et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Abraham, C. & Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 15, 1090–1100 (2009).
Cario, E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm. Bowel Dis. 16, 1583–1597 (2010).
Franchi, L., Munoz-Planillo, R. & Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 (2012).
Fritz, T., Niederreiter, L., Adolph, T., Blumberg, R.S. & Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 60, 1580–1588 (2011).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Okazaki, T. et al. Contributions of IBD5, IL23R, ATG16L1, and NOD2 to Crohn’s disease risk in a population-based case–control study: evidence of gene–gene interactions. Inflamm. Bowel Dis. 14, 1528–1541 (2008).
Glocker, E.O., Kotlarz, D., Klein, C., Shah, N. & Grimbacher, B. IL-10 and IL-10 receptor defects in humans. Ann. N Y Acad. Sci. 1246, 102–107 (2011).
Coccia, M. et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 209, 1595–1609 (2012).
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993).
Hugot, J.P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 (2001).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606 (2001).
Villani, A.C. et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Siegmund, B., Lehr, H.A., Fantuzzi, G. & Dinarello, C.A. IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA 98, 13249–13254 (2001).
Ligumsky, M., Simon, P.L., Karmeli, F. & Rachmilewitz, D. Role of interleukin 1 in inflammatory bowel disease-enhanced production during active disease. Gut 31, 686–689 (1990).
Reinecker, H.C. et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn's disease. Clin. Exp. Immunol. 94, 174–181 (1993).
Casini-Raggi, V. et al. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J. Immunol. 154, 2434–2440 (1995).
McAlindon, M.E., Hawkey, C.J. & Mahida, Y.R. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 42, 214–219 (1998).
Henao-Mejia, J., Elinav, E., Strowig, T. & Flavell, R.A. Inflammasomes: far beyond inflammation. Nat. Immunol. 13, 321–324 (2012).
Rathinam, V.A., Vanaja, S.K. & Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 13, 333–332 (2012).
Papatriantafyllou, M. Mucosal immunology: inflammasome shapes the microbiota. Nat. Rev. Immuno.l 11, 439 (2011).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Allen, I.C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Hirota, S.A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Gross, O., Thomas, C.J., Guarda, G. & Tschopp, J. The inflammasome: an integrated view. Immunol. Rev. 243, 136–151 (2011).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Sarra, M., Pallone, F., Macdonald, T.T. & Monteleone, G. IL-23/IL-17 axis in IBD. Inflamm. Bowel Dis. 16, 1808–1813 (2010).
Sutton, C.E. et al. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31, 331–341 (2009).
Caprioli, F., Pallone, F. & Monteleone, G. Th17 immune response in IBD: a new pathogenic mechanism. J. Crohns Colitis 2, 291–295 (2008).
Mills, K.H., Dungan, L.S., Jones, S.A. & Harris, J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J. Leukoc. Biol. 93, 489–497 (2013).
Murai, M. et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 10, 1178–1184 (2009).
Zhang, L., Yuan, S., Cheng, G. & Guo, B. Type I IFN promotes IL-10 production from T cells to suppress Th17 cells and Th17-associated autoimmune inflammation. PLoS One 6, e28432 (2011).
Zaki, M.H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Vijay-Kumar, M. et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest. 117, 3909–3921 (2007).
Carvalho, F.A. et al. Interleukin-1beta (IL-1beta) promotes susceptibility of Toll-like receptor 5 (TLR5) deficient mice to colitis. Gut 61, 373–384 (2012).
Reuter, B.K. & Pizarro, T.T. Commentary: the role of the IL-18 system and other members of the IL-1R/TLR superfamily in innate mucosal immunity and the pathogenesis of inflammatory bowel disease: friend or foe? Eur. J. Immunol. 34, 2347–2355 (2004).
Chang, E.Y., Guo, B., Doyle, S.E. & Cheng, G. Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J. Immunol. 178, 6705–6709 (2007).
Guarda, G. et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223 (2011).
Inoue, M. et al. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 5, ra38 (2012).
Katakura, K. et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 115, 695–702 (2005).
Kole, A. et al. Type I IFNs regulate effector and regulatory T cell accumulation and anti-inflammatory cytokine production during T cell-mediated colitis. J. Immunol. 191, 2771–2779 (2013).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Ivanov, I.I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Kosiewicz, M.M. et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J. Clin. Invest. 107, 695–702 (2001).
Guo, B., Chang, E.Y. & Cheng, G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118, 1680–1690 (2008).
Liu, B. et al. Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc. Natl. Acad. Sci. USA 110, 6877–6882 (2013).
Acknowledgements
This work was supported by NIH grants AI070603, AI077283, and HL100556 (to Z.L.), and NIAID K22 AI87707 (to B.G.). We thank members of our laboratory for their discussion and suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Zhang, J., Fu, S., Sun, S. et al. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol 7, 1139–1150 (2014). https://doi.org/10.1038/mi.2014.1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.1
This article is cited by
-
hucMSC-derived exosomes attenuate colitis by regulating macrophage pyroptosis via the miR-378a-5p/NLRP3 axis
Stem Cell Research & Therapy (2021)
-
Apoptosis inhibitor of macrophage (AIM) contributes to IL-10-induced anti-inflammatory response through inhibition of inflammasome activation
Cell Death & Disease (2021)
-
Genotype–phenotype associations of polymorphisms within the gene locus of NOD-like receptor pyrin domain containing 3 in Swiss inflammatory bowel disease patients
BMC Gastroenterology (2021)
-
The Influence of Bifidobacterium bifidum and Bacteroides fragilis on Enteric Glial Cell–Derived Neurotrophic Factors and Inflammasome
Inflammation (2020)
-
MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice
Scientific Reports (2018)
