
Abstract
Toll-like receptor-5 (TLR5)-mediated detection of flagellin induces nuclear factor (NF)-κB-mediated transcription of host defense gene expression, whereas recognition of intracellular flagellin by interleukin (IL)-1-converting enzyme protease-activation factor (IPAF) results in maturation/secretion of the inflammasome cytokine IL-1β. The potent effects of IL-1β are counter-regulated by secretory IL-1 receptor antagonist (sIL-1Ra). We studied the roles of flagellin receptors in regulating the expression of IL-1β and sIL-1Ra and their subsequent roles in inflammation. Flagellin induced sIL-1Ra in intestinal epithelia and macrophages in a dose- and time-dependent manner, whereas IL-1β was only induced in macrophages. In vivo, flagellin-induced sIL-1Ra, but not IL-1β, was absolutely dependent upon TLR5 expressed on non-hemopioetic cells. Thus, loss of TLR5 increased the IL-1β/sIL-1Ra ratio on flagellin treatment, which correlated with increased inflammatory pathology in response to this product. Furthermore, the flagellin/TLR5 interaction was important for the induction of sIL-1Ra and limiting inflammatory pathology on Salmonella infection. Finally, reduced sIL-1Ra levels in TLR5KO mice correlated with spontaneous colitis. Taken together, we demonstrate that intestinal epithelia, despite not expressing IL-1β, secrete sIL-1Ra in a TLR5-dependent manner suggesting that loss of TLR5 may promote inflammation by increasing IL-1β activity. Thus, optimizing the balance between inflammasome cytokines and their endogenous inhibitors might prove a useful strategy to treat inflammatory disorders.
Similar content being viewed by others


Introduction
The mammalian innate immune system is composed of numerous highly conserved pattern recognition receptors. These include toll-like receptors (TLRs) and nod-like receptors that recognize specific microbial components. This recognition leads to an innate immune response and finally culminates in the recruitment of professional immune cells to the site of infection. Microbial-derived molecules detected by TLRs include peptidoglycan, double-stranded RNA, lipopolysaccharide (LPS), flagellin, and CpG DNA, which are recognized by TLRs 2, 3, 4, 5, and 9, respectively.1, 2 Such TLR-mediated detection of LPS, peptidoglycan, and flagellin is mediated at the cell surface. Some of these molecules and/or their subunits are also recognized by nod-like receptors; for example, peptidoglycan-derived muramyl-dipeptide is recognized intracellularly by Nod1 and Nod2, whereas cytoplasmic flagellin, the primary component of flagella, is recognized by interleukin (IL)-1-converting enzyme protease-activation factor (IPAF, also known as NLRC4) and nod-like receptor apoptosis-inhibitory protein-5.3, 4, 5 At least in mice, TLR5 is not highly expressed by most populations of macrophages and dendritic cell, but rather is functionally expressed on non-hemopoietic cells, particularly epithelial cells.6 Ligation of TLR5 by flagellin results in nuclear factor (NF)-κB-mediated induction of a number of cytokines, including keratinocyte derived chemokine (KC), IL-6, and granulocyte-colony-stimulating factor, while also promoting cell survival. Such TLR5-mediated signaling results in the recruitment/activation of immune cells to the site of infection and prepares resident cells to withstand the stress created by the bacterium and recruited immune cells. In contrast to TLR5-mediated detection of extracellular flagellin, IPAF and nod-like receptor apoptosis-inhibitory protein-5-mediated recognition of cytosolic flagellin is preferential, or perhaps exclusive, to macrophages. Moreover, the primary consequence of this signaling pathway is activation of the inflammasome complex. Such inflammasome activation results in the caspase-1-mediated cleavage of pro-IL-1β and pro-IL-18 into their mature active forms; hence, IL-1β and IL-18 are often referred to as inflammasome cytokines. The accumulated data indicate that TLR5 activation results in the induction of cell survival genes, whereas IPAF activation leads to pyroptosis, a form of necrotic cell death.7, 8
Use of an additional checkpoint to regulate the production of inflammasome cytokines likely reflects the importance of preventing excessive promotion of inflammation by these cytokines. This notion is underscored by the fact that the biological activity of inflammasome cytokines is further regulated by expression of endogenous soluble inhibitor proteins. Indeed, the absence of induction of these anticytokines results in unchecked inflammation that causes tissue damage to the host.9 The best studied example of counter regulation of inflammasome cytokines, and the focus of this article, is the secretory IL-1 receptor antagonist (sIL-1Ra), which competes with IL-1β for its receptor, thus dampening the bioactivity of this potent proinflammatory cytokine. sIL-1Ra expression can be induced by proinflammatory stimuli, such as IL-1β itself and LPS, as well as by a variety of immunomodulators such as granulocyte–macrophage-colony-stimulating factor, interferon (IFN)-β, and IFN-γ.10, 11 Importantly, sIL-1Ra is much more broadly expressed than IL-1β, allowing cells that do not make IL-1β to have a role in regulating its activity.
Mice lacking the gene for sIL-1Ra are highly susceptible to septic shock and predisposed to spontaneous inflammatory disorders, underscoring the importance of sIL-1Ra in host health.12, 13 Interestingly, such mice are resistant to Listeria, highlighting the close relationship between inflammation and host defense.14 With regard to these, mice lacking sIL-1Ra have some phenotypic similarities to mice lacking TLR5, as these are also prone to inflammation and are relatively resistant to Salmonella infection.15, 16 Thus, we hypothesized that the flagellin/TLR5 interaction might result in induction of sIL-1Ra and interference with this interaction might promote inflammation. Herein, we observed that flagellin-induced TLR5-mediated sIL-1Ra has a key role in attaining the proper balance of sIL-1Ra and IL-1β and, consequently, its reduced levels may have a role in the excessive and/or spontaneous inflammation that results from loss of the flagellin/TLR5 interaction.
Results
Flagellin induces sIL-1Ra in intestinal epithelial cells
Inflammasome cytokines, especially IL-1β, are increasingly recognized as being key mediators of inflammation. Thus, regulation of expression of IL-1β and the endogenous inhibitor of IL-1β, sIL-1Ra, may have a key role in determining the extent to which an immune-mediated inflammatory response is properly initiated and then terminated. The ability to assemble functional inflammasome complexes and, consequently, produce mature IL-1β appears to be limited to select populations of hemopoietic cells, particularly macrophages and dendritic cells. In accordance, we recently showed that TLR5, known to be preferentially expressed on intestinal epithelial cells (IECs) and other non-hemopoietic cells, was not required for flagellin-induced production of inflammasome cytokines.17, 18 However, the ability to produce inhibitors of inflammasome cytokines appears to be considerably more widespread, possibly reflecting a divergent role for these proteins in limiting inflammation. Thus, we hypothesized that bacterial flagellin, which is thought to be a dominant innate immune activator of IEC, might induce these cells to produce negative regulators of inflammasome cytokines. In support of this hypothesis, flagellin induced robust production of sIL-1Ra from IEC in a time- and dose-dependent manner (Figure 1a and b). Such IEC production of sIL-1Ra was independent of IL-1β, in which, in accordance with other studies, IECs failed to produce detectable levels of IL-1β (IEC production of IL-1β was below the limit of detection in all conditions examined).17, 19 The time course of sIL-1Ra secretion in response to flagellin treatment was similar to that observed for IL-8 secretion (Figure 1c), suggesting that sIL-1Ra might be one of the many NF-κB-regulated genes, in which expression is induced in IEC on exposure to flagellin. To test this possibility, IEC were exposed to a proteasome inhibitor that is commonly used in vitro to block NF-κB activation. As in our previous work,20 this treatment completely blocked flagellin-induced IL-8 secretion (Figure 1d). Such NF-κB blockade significantly reduced but did not eliminate flagellin-induced sIL-1Ra production, suggesting that NF-κB is but one of a number of signaling pathways induced by flagellin that can eventuate in sIL-1Ra expression in IEC (Figure 1e). In accordance, induction of sIL-1Ra expression was not restricted to agonists that primarily activate NF-κB but was observed in response to type I IFN (Figure 1f), as well as type II IFN, which is known to induce inhibitors of inflammasome cytokines in IEC and thus served as a positive control.21, 22 Induction of sIL-1Ra was not restricted to proinflammatory agonists as epidermal growth factor, which did not elicit detectable IL-8 secretion23 (data not shown), elicited modest but yet significant induction of sIL-1Ra (Figure 1g). Thus, IEC can make sIL-1Ra in response to a variety of agonists, including low concentrations of flagellin.

Secretion of secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) by intestinal epithelial cells (IECs). Confluent human model intestinal epithelia (HT29) were stimulated with indicated doses of flagellin (FliC), epidermal growth factor (EGF) or 100 IU/ml of human interferon (IFN)α, IFNβ or IFNγ. Supernatants were taken after 24 h of culture or at indicated time periods for IL-8 or sIL-1Ra analysis by enzyme-linked immunosorbent assay. (a) Dose-dependent secretion of sIL-1Ra. (b) sIL-1Ra and (c) IL-8 time-dependent secretion after stimulation with 100 ng/ml of FliC (triangle) or control phosphate-buffered saline (circle). (d) IL-8 and (e) sIL-1Ra secretion after cells were preincubated for 30 min with dimethylsulfoxide (DMSO) or MG262 in DMSO (10 nM) and then stimulated with 100 ng/ml of FliC. (f) Induction of sIL-1Ra by type I and II IFNs. (g) Induction of sIL-1Ra by EGF. *P<0.05.
Next, we examined in the context of an in vitro bacterial infection the extent to which flagellin had an important role in mediating IEC production of sIL-1Ra. IECs were infected with wild-type (WT) Salmonella enterica serovar Typhimurium or an isogenic flagellin-deficient mutant. In accordance with our previous works that suggest flagellin is absolutely required for S. Typhimurium-induced NF-κB activation,24 loss of S. Typhimurium flagellin markedly attenuated IEC production of IL-8 in response to this bacterium (Figure 2a). In contrast, both WT and aflagellate Salmonella induced significant production of sIL-1Ra but, nonetheless, the level of IEC production of sIL-1Ra was significantly less in response to the aflagellate strain (Figure 2b). In accordance with the notion that IEC lack the ability to produce inflammasome cytokines, neither Salmonella strain elicited detectable IL-1β production by IEC (data not shown). Thus, although a number of signaling pathways can induce IEC to secrete sIL-1Ra, the flagellin/TLR5 interaction in IEC detection can be envisaged to perhaps have a significant role in negatively regulating IL-1β activity.

Flagellin deficiency attenuates Salmonella's ability to induce sIL-1Ra in intestinal epithelial cells. Confluent human intestinal epithelial cells (HT29) were exposed to 1 × 108 colony-forming unit/ml of either flagellate S. Typhimurium or its isogenic aflagellate mutant. After 3 h of infection, cells were washed three times and incubated with medium containing 50 μg/ml of gentamicin for 6 h. (a) Interleukin (IL)-8 and (b) secretory IL-1 receptor antagonist (sIL-1Ra) were measured in culture supernatant by enzyme-linked immunosorbent assay. *P<0.05.
Flagellin-induced IL-1β and sIL-1Ra in macrophages is TLR5-independent
Most populations of murine macrophages have been reported to lack functional expression of TLR5.6, 25 However, macrophages exhibit IPAF-mediated production of mature IL-1β on detection of cytosolic flagellin. As IL-1β itself is known to elicit production of sIL-1Ra from a variety of cell types, we predicted flagellin would also induce sIL-1Ra in macrophages. We observed that, indeed, flagellin- and LPS-stimulated macrophages (J774.A1) secreted significant amounts of sIL-1Ra in comparison with unstimulated macrophages (Figure 3a). The level of sIL-1Ra elicited by flagellin was only modestly less than that induced by LPS, suggesting the possibility that flagellin may have a significant role in regulating macrophages expression of inflammasome cytokines in vivo. Similar to IEC, macrophages produced sIL-1Ra in response to flagellin in a dose- and time-dependent manner (Figure 3b and c). The time course of flagellin-induced sIL-1Ra expression by macrophages was relatively slow in accordance with it being mediated by intracellular detection. In agreement with recent studies, flagellin-induced IL-1β secretion by macrophages was IPAF-dependent and TLR5-independent (Figure 3d). A similar pattern was observed for flagellin-induced macrophage production of sIL-1Ra, in which it was also TLR5-dependent and partially dependent on IPAF (Figure 3e).

Secretion of secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) by murine macrophages. Confluent mouse macrophage cell line J774A.1 were grown in 24-well plate and stimulated with indicated doses of flagellin (FliC), 20 ng of lipopolysaccharide (LPS) or 100 IU/ml of mouse interferon (IFN)γ. Supernatants were collected after 24 h of culture or at indicated time periods for IL-1β or sIL-1Ra analysis by enzyme-linked immunosorbent assay (ELISA). (a) sIL-1Ra production by murine macrophages after stimulation with FliC (10 μg/ml), mouse IFNγ (100 IU/ml) or LPS (20 ng/ml). (b) Dose-dependent and (c) time-dependent secretion of sIL-1Ra by J774A.1 cells after stimulation with flagellin. (d) IL-1β and (e) sIL-1Ra production in response to FliC (10 μg/ml) by resident peritoneal wild-type (WT), TLR5-deficient (T5KO) and IL-1-converting enzyme protease-activation factor (IPAF)-deficient (IPAFKO) macrophages measured by ELISA. *P<0.05.
We next examined whether flagellin had a significant role in induction of sIL-1Ra on encountering S. Typhimurium. As, in contrast to IEC, macrophages respond robustly to a variety of TLR ligands, notably LPS and CpG DNA, it seemed likely that flagellin might be dispensable for eliciting proinflammatory gene expression from these cells. In accordance, WT and aflagellate S. Typhimurium-infected macrophages produced similar amounts of tumor necrosis factor (TNF)α, showing that loss of flagellin did not have a negative impact on TNFα production, a commonly used readout for macrophage activation (Figure 4a). In contrast, loss of flagellin from S. Typhimurium significantly reduced its ability to elicit both IL-1β and sIL-1Ra (Figure 4b and c). As the reduction in levels of IL-1β and sIL-1Ra were similar, absence of S. Typimurium flagellin caused only a modest change in the IL-1β/sIL-1Ra ratio (Figure 4d). Thus, when taken together, our in vitro and ex vivo results suggest that recognition of flagellin by both IEC TLR5 and macrophage IPAF might contribute to sIL-1Ra expression, while loss of the IEC sIL-1Ra production, which occurs without concomitant loss of IL-1β, might have a greater potential to shift the ratio of IL-1β/sIL-1Ra.26

Reduced induction of interleukin (IL)-1β and secretory IL-1 receptor antagonist (sIL-1Ra) by flagellin-deficient Salmonella in macrophages. Confluent mouse macrophages were grown in 24-well plate (2 × 105 cells/well) and exposed to 1 × 108 colony-forming unit/ml of either flagellate S. Typhimurium or its isogenic aflagellate mutant. After 1 h of infection, cells were washed three times and incubated with medium containing 50 μg/ml of gentamicin for 24 h. Supernatants were assayed for (a) tumor necrosis factor (TNF)α, (b) IL-1β and (c) sIL-1Ra by enzyme-linked immunosorbent assay. (d) The IL-1β activity was determined by calculating the ratio of IL-1β/sIL-1Ra. *P<0.05.
Flagellin-induced sIL-1Ra is TLR5-dependent in vivo
The above-described studies suggested that loss of TLR5 in vivo might alter the relative balance of IL-1β and sIL-1Ra. To begin to investigate this possibility, we systemically exposed mice (WT, TLR5KO, IPAFKO and TLR5/IPAF double knockout) to flagellin intraperitoneally (i.p.). This treatment resulted in significant rapid elevation of serum sIL-1Ra, KC and IL-6 that was predominantly dependent on TLR5 (Figure 5a–c) and did not have a clear requirement for IPAF. In accordance with previous work, flagellin did not induce a detectable increase in serum IL-1β. Such systemic treatment with flagellin also increased the level of sIL-1Ra, KC and IL-6 in the intestine, in which a significant elevation in IL-1β could also be measured (Figure 5d–g). Analogous to serum, flagellin-induced colonic production of sIL-1Ra, KC and IL-6 exhibited a near-total dependence on TLR5. In contrast, loss of TLR5 reduced but did not eliminate flagellin-induced IL-1β (Figure 5g) with the reduction likely reflecting the requirement for TLR5 to induce IL-1β mRNA (Figure 5h). In WT mice, the relative increase in colonic IL-1β occurred concomitant with an increase in sIL-Ra, such that flagellin treatment resulted in a decrease in the IL-1β/sIL-1Ra ratio in WT mice in comparison with untreated mice (Figure 5i). In contrast, flagellin treatment increased the ratio of IL-1β/sIL-1Ra in TLR5-deficient mice. Such an increase in the IL-1β/sIL-1Ra ratio was not observed in mice lacking IPAF or both IPAF/TLR5 after flagellin treatment. These results support the notion that loss of TLR5 has the potential to result in an elevated IL-1β/sIL-1Ra ratio in vivo.

Flagellin induced secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) is toll-like receptor-5 (TLR5)-dependent in vivo. Six- to eight-week-old wild-type (WT), TLR5-deficient (T5KO), IL-1-converting enzyme protease-activation factor (IPAF)-deficient (IPAFKO) or IPAF/TLR5 (double knockout (DKO)) mice (n=5) were administered intraperitoneally (i.p.) 200 μl of phosphate-buffered saline (PBS) or 10 μg FliC/mouse in 200 μl of PBS. After 1 h, serum was collected and assayed for (a) sIL-1Ra, (b) KC and (c) IL-6 by enzyme-linked immunosorbent assay (ELISA). Colons were cultured for 24 h and (d) sIL-1Ra, (e) KC, (f) IL-6 and (g) IL-1β in supernatants were assayed by ELISA. (h) Total RNAs from colon were isolated and Pro-IL-1β mRNA levels were measured by quantitative reverse transcription-PCR. (i) IL-1β/sIL-1Ra ratio. The data are representative of three independent experiments. *P<0.05.
Flagellin-induced sIL-1Ra is non-hemopoietic cell-dependent in vivo
Next we examined the relative contribution of hemopoietic (radiosensitive) and non-hemopoietic (radioresistant) cells in inducing sIL-1Ra in response to systemic flagellin. We generated bone marrow chimeric mice (WT → WT, WT → TLR5KO, TLR5KO → TLR5KO, and TLR5KO → WT) as described in Methods section by whole body γ-radiation. We have found that this regimen consistently results in a level of chimerism of 95–99% at 8 weeks after the procedure.27 Bone marrow chimeric mice were given either 0.1 ml of phosphate-buffered saline or flagellin (10 μg/mouse in 0.1 ml PBS) i.p. and after 1 h analyzed for serum sIL-1Ra levels by enzyme-linked immunosorbent assay (ELISA). As shown in Figure 6, mice with TLR5 in their non-hemopoietic cells (i.e., WT → WT and TLR5KO → WT chimeras) induced significant levels of serum sIL-1Ra in response to systemic treatment with flagellin, whereas mice lacking TLR5KO in their non-hemopoietic cells (i.e., TLR5KO → TLR5KO and WT → TLR5KO) did not exhibit induction of sIL-1Ra in response to flagellin, regardless of whether their hemopoiteic cells had the TLR5 gene. These results suggest that TLR5 expressed in non-hemopoietic cells mediates the induction of sIL-1Ra in response to flagellin.

Flagellin induced secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) is dependent on non-hemopoietic cells in vivo. Seven- to eight-week-old wild-type (WT) or TLR5KO mice were irradiated and generated WT → WT, WT → TLR5KO, TLR5KO → TLR5KO, and TLR5KO → WT bone marrow chimeras were generated as described in Methods section. After 8 weeks, they were challenged with either phosphate-buffered saline or flagellin (10 μg/mouse) intraperitoneally, and after 1 h blood was collected and serum sIL-1Ra levels were analyzed by enzyme-linked immunosorbent assay. *P<0.05.
Loss of TLR5 alters flagellin-induced IL-1β/sIL-1Ra ratio and correlates with inflammation in vivo
To examine the potential consequences of such loss of sIL-1Ra in TLR5KO mice, WT and TLR5KO mice were given 10 injections of flagellin i.p. over a course of 20 days. This treatment did not have a marked effect on WT mice, but resulted in weight loss in TLR5-deficient mice (Figure 7a). Moreover, flagellin-treated TLR5KO mice displayed evidence of intestinal inflammation including elevated levels of neutrophil marker myeloperoxidase (MPO), sporadic lymphocytic infiltrates and loss of crypts. (Figure 7b–d). These results suggest that loss of flagellin-induced sIL-1Ra production might result in potentiated or prolonged inflammation.

Flagellin administration induces adverse effects in toll-like receptor-5 (TLR5)-deficient mice. Six- to eight-week-old wild-type (WT) or TLR5-deficient (T5KO) mice were administered either 100 μl of phosphate-buffered saline (PBS) or flagellin (FliC; 25 μg/mouse) in 100 μl PBS on alternate days over a period of 20 days. (a) At the end of the experiment, relative change in body weight between the first and the tenth injection was calculated. (b) Colonic myeloperoxidase (MPO) and (c) cecum MPO. (d) Hematoxylin and eosin-stained colon from mice that received FliC. The results are representative of two independent experiments (n=8). *P<0.05.
We next sought to investigate the role of intestinal TLR5-induced sIL-1Ra in response to an infectious challenge. We and others have shown that flagellin-deficient S. Typhimurium is more virulent than its isogenic flagellated S. Typhimurium and results in greater inflammatory pathology in the intestine in Balb/c mice.7, 28, 29 Herein, we observed that such enhanced inflammatory pathology correlated with loss of sIL-1Ra, in that flagellate S. Typhimurium, but not an aflagellate isogenic mutant, induced significant serum sIL-1Ra production in Balb/c mice (Figure 8a). The increased pathology that resulted from loss of flagellin can be phenocopied in C57BL/6 mice by deletion of murine TLR5.16 Here, we observed that loss of TLR5 but not IPAF reduced sIL-1Ra production in response to flagellate S. Typhimurium (Figure 8b). All four genotypes of C57BL/6 mice (WT, TLR5KO, IPAFKO and IPAF/TLR5 double knockout) failed to exhibit elevations in IL-1β or sIL-1Ra on challenge with flagellin-deficient S. Typhimurium (data not shown). Thus, loss of sIL-1Ra production may have a role in mediating the severe inflammation observed, which occurs in response to S. Typhimurium infection in the absence of the flagellin/TLR5 interaction.

Salmonella flagellin and toll-like receptor-5 interaction is required for in vivo secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) secretion. (a) Eight-week-old BALB/CJ mice were pretreated with streptomycin, and 24 h later infected orally with flagellate S. Typhimurium or its aflagellate isogenic mutant. 48 h post-infection, mice were bled and serum analyzed by enzyme-linked immunosorbent assay (ELISA) for sIL-1Ra levels. (b) Eight-week-old wild-type (WT), TLR5KO (T5KO), IPAFKO and IPAF/TLR5 DKO mice were orally infected with flagellate S. Typhimurium or its aflagellate isogenic mutant (1 × 108 colony-forming unit/mouse). Twelve hours post-infection, mice were bled and serum sIL-1Ra quantified by ELISA. The results are representative of two independent experiments. *P<0.05.
Finally, we considered the extent to which loss of sIL-1Ra might correlate with the spontaneous colitis exhibited by some TLR5-deficient mice. Specifically, we previously showed that loss of TLR5 also predisposed mice to developing spontaneous colitis, the severity of which correlates with levels of serum amyloid A.15 Here, we observed that levels of serum amyloid A are inversely correlated with serum levels of sIL-1Ra (Figure 9). This suggests that loss of IEC production of sIL-1Ra resulting in elevated levels of IL-1β activity may have a role in the spontaneous colitis exhibited by TLR5-deficient mice.

*Figure 9 Systemic secretory interleukin (IL)-1 receptor antagonist (sIL-1Ra) inversely correlates with severity of spontaneous colitis in toll-like receptor-5 (TLR5)-deficient mice. Eight-week-old wild-type (WT) or TLR5-deficient (T5KO) mice with low serum amyloid A (SAA) levels (mild colitis), with high SAA levels (robust colitis), or with rectal prolapse (severe colitis) were bled and serum sIL-1Ra levels were measured by enzyme-linked immunosorbent assay. *P<0.05.
Discussion
As the first and most potent endogenous pyrogen ever identified, IL-1β has long been appreciated as a key mediator in a variety of disease states associated with inflammation.30 Relative to other endogenous mediators of inflammation, regulation of IL-1β activity is quite complex likely reflecting that stringent control is needed to prevent this potent cytokine from driving excessive inflammation. Specifically, in addition to being regulated at the transcriptional level by proinflammatory transcription factors, such as NF-κB that are themselves tightly regulated, the rate-limiting step of IL-1β expression is inflammasome-mediated caspase-1 cleavage of pro-IL-1β into its mature bioactive form.31 Additionally, once secreted, IL-1β activity is regulated by the production of sIL-1Ra, which competes with IL-1β for IL-1R binding. In the absence of inflammation, sIL-1Ra is typically present in about 10-fold molar excess of IL-1β.32, 33 Importantly, the ability of cells to make sIL-1Ra is much more widespread than is the case for IL-1β, and thus cells that do not make IL-1β may, nonetheless, have a pivotal role in regulating IL-1β activity.34 Herein, we show that, in vitro and in vivo, IECs are an example of this concept as they make robust levels of sIL-1Ra but not IL-1β, and thus may have a key role in keeping IL-1β activity in check. In support of this notion, on injection of purified flagellin, loss of TLR5-mediated sIL-1Ra resulted in IPAF-mediated IL-1β production, causing a net increase in the IL-1β/sIL-1Ra ratio, whereas this metric was decreased in WT mice. Such alteration in flagellin-induced IL-1β/sIL-1Ra ratio, herein referred to as IL-1β activity, correlated with the occurrence of flagellin-induced inflammatory pathology that was observed in TLR5KO, but not WT, mice. Given the established role of IL-1β in a number of proinflammatory disease states, this correlation suggests that loss of TLR5-mediated sIL-1Ra production may have a role in the tendency of TLR5-deficient mice to develop inflammation. However, correlation does not establish causation and thus, future experiments, likely with IL-1R-deficient mice will be necessary to directly investigate the role of IL-1β in the susceptibility of TLR5KO mice to inflammation.
IL-1β itself is one potent inducer of sIL-1Ra, thus providing a “built-in” mechanism of counter-regulation, wherein increased production of IL-1β will induce sIL-1Ra to curtail IL-1β activity.10 This mechanism is operable in mouse macrophages as flagellin-induced IPAF-mediated IL-1β production was necessary for these cells to produce sIL-1Ra. Built-in inhibitory mechanisms are common in proinflammatory signaling in general with an important example being cytokine-induced NF-κB activation inducing expression of I-κB that shuts down NF-κB. In addition, sIL-1Ra can also be made independent of IL-1β with, for example, IL-1β-deficient mice producing high levels of sIL-1Ra.35 Thus, regulation of IL-1β activity is also controlled by “free-standing” signaling pathways. In accordance, IEC expression of sIL-1Ra was upregulated by flagellin, which activates the canonical NF-κB pathway but was also induced by type I and II IFNs and even the epidermal growth factor that do not activate proinflammatory signaling cascades, such as NF-κB and p38 MAPK. Thus, in vivo, we envisage that IEC may be integrating a variety of different signals to contribute to the regulation of IL-1β bioactivity. Accordingly, our results with bone marrow chimeric mice clearly demonstrate that flagellin-induced sIL-1Ra was dependent on TLR5 expressed in non-hemopioetic cells. Nonetheless, loss of one of the major signaling pathways, namely, flagellin-induced TLR5 signaling, governing S. Typhimurium intestinal interaction was sufficient to reduce IEC production of sIL-1Ra and result in increased IL-1β activity in vitro and in vivo, indicating that loss of select signaling pathways may significantly increase IL-1β activity without necessarily increasing levels of IL-1β.
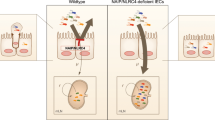
Loss of the TLR5/flagellin interaction makes mice lacking TLR5 more prone to developing inflammation.15 Specifically, aflagellate S. Typhimurium induces more severe inflammatory pathology than flagellate S. Typhimurium, whereas TLR5KO exhibit more severe colitis in response to this pathogen.16 Moreover, TLR5KO mice can develop spontaneous colitis or metabolic syndrome and exhibit more severe colitis on exposure to dextran sulfate sodium.15, 36, 37 Much of this hyperinflammatory phenotype is thought to result from excess activation of other TLR signaling pathways due to inability to efficiently clear perturbing microbes. This notion is supported by the observation that some aspects of this phenotype are absent in mice lacking both TLRs 4 and 5, while all are absent in MyD88KO mice, which are broadly deficient in TLR signaling.15, 38 However, it is now well-appreciated that MyD88-dependent phenotypes can reflect dependency on IL-1β (or IL-18) rather than, and/or in addition to, TLR signaling. Thus, the results herein suggest that loss of non-hemopioetic TLR5-mediated sIL-1Ra production may contribute to the hyperinflammatory phenotype in TLR5KO mice. In accordance, even in the absence of a challenge, TLR5KO mice exhibited a reduction of sIL-1Ra expression with the extent of reduction correlating with the severity of their spontaneous inflammation in contrast to another animal model of colitis.39 This correlation makes clear that loss of TLR5 is not the only means of reducing sIL-1Ra expression (as colitic and non-colitic TLR5KO mice are all completely deficient in TLR5), but it does suggest that loss of TLR5-induced sIL-1Ra may be a significant factor in tipping the balance toward inflammation in these mice (Figure 10). Thus, strategies to treat/prevent chronic inflammatory disorders might consider attempting to optimize the balance between inflammasome cytokines and their endogenous inhibitors.

Schematic representation of role of IL-Iβ in TLR5 deficient phenotype (See Discussion).
Methods
Cell culture. Model IECs (HT29) and mouse macrophage cell line J774A.1 (purchased from ATCC, Manassas, VA) were grown to confluence in a 24-well plate (5 × 105 cells/well) using Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 1% penicillin and streptomycin and non-essential amino acids. On the day of stimulation, cells were washed twice with PBS and stimulated with flagellin, LPS (Sigma, St Louis, MO), epidermal growth factor (R&D Systems, Minneapolis, MN), IFNα, IFNβ, or IFNγ (National Institute of Allergy and Infectious Diseases Reference Reagent Laboratory, supplied by ATCC) in serum-free DMEM for indicated time points. For indicated experiments, cells were preincubated for 30 min with 10 nM of MG262 inhibitor (Sigma) in dimethylsulfoxide. Control cells were incubated with equal amounts of dimethylsulfoxide alone. The supernatants were collected, centrifuged and stored at −20°C until analysis. Clear supernatants from HT29 were used for IL-8, IL-1β and sIL-1Ra, whereas supernatants from macrophages (J774A.1) were used for assaying TNFα, IL-1β, and sIL-1Ra by ELISA.
In vitro Salmonella infection. WT S. enterica serovar Typhimurium (SL3201) phase variable (WT) and its isogenic mutant (aflagellate, phenotype: nonmotile; genotype: fliC–/fljB–) were grown under microaerophilic conditions as previously described.7 HT29 cells and macrophages were exposed to indicated bacterial strain using a modified gentamicin protection assay.40, 41 Briefly, HT29 and macrophages were seeded in 24-well tissue culture plates with 2 × 105 cells/well, respectively. Monolayers were then exposed to 1 × 108 colony-forming unit/ml without antibiotics. After a 3 h incubation period for HT29 cells or 1 h for macrophages at 37°C, monolayers were washed three times in PBS and incubated with fresh culture medium containing 50 μg/ml of gentamicin (Sigma), and supernatants were collected (at 6 h for HT29 cells and for 24 h for macrophages), centrifuged and stored at −20°C until analysis for cytokines.
Peritoneal exudates cell. Resident peritoneal macrophages were collected from killed animals by DMEM lavage, resuspended in DMEM containing 10% fetal bovine serum and seeded at the density of 4 × 105 cells/well. After overnight incubation, cells were washed to remove non-adherent cells and stimulated with indicated stimuli in serum-free media.
Mice. Six- to eight-week-old WT, TLR5KO, IPAFKO and IPAF/TLR5 double knockout mice under C57BL/6 background (backcrossed to C57BL/6 mice for 10 generations) were bred and maintained in Emory University animal facility. Eight-week-old female BALB/CJ mice were purchased from Jackson Laboratories (Bar Harbar, ME). All experiments involving animals were approved by the Emory University animal ethical committee and performed according to legal requirements.
Generation of bone marrow chimeric mice. Femurs from donor mice were flushed with DMEM supplemented with 10% fetal bovine serum and erythrocytes were lysed using BD Pharm Lyse lysis buffer (BD Biosciences Pharmingen, San Jose, CA). Recipient mice were exposed to 11 Gy whole body γ-radiation using a Gammacell 40 137Cs irradiator (Model GC-40, Ontario, Canada) and then given 1 × 107 bone marrow cells in HBSS through retro-orbital plexus from the appropriate donor strain. All recipients were females between 7 and 8 weeks of age. Immediately after transfer, mice were placed on drinking water containing 2 mg/ml neomycin sulfate (Mediatech, Manassas, VA) for 2 weeks. Mice were allowed to rest 8 weeks before experimental use. This procedure yields 95–99% chimerism as previously described by Sanders et al.27
Flagellin administration. Flagellin (FliC) from WT S. enterica serovar Typhimurium (SL3201, fljB–) was purified through sequential cation and anion-exchange chromatography and purity was verified as previously described.20 Mice were administered either 0.1 ml PBS or flagellin (10 μg/mouse in 0.1 ml PBS) i.p. and bled after 1 h by retro-orbital plexus, and hemolysis-free serum was collected in serum separator tubes (Becton and Dickinson, Franklin Lakes, NJ). To study flagellin's adverse effects, 4-week-old WT and TLR5KO mice were given 10 injections of PBS or flagellin (25 μg/mouse) i.p. on alternate days. Body weights were monitored every day. Mice were killed by CO2 asphyxiation and tissues were collected for MPO assay and histology.
Quantitative RT-PCR. Total RNA were isolated from colonic tissues using TRIzol (Invitrogen, Carlsbad, CA) and was purified using the RNeasy Plus Mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA was quantified in realplex2 (Eppendorf, Hauppauge, NY) using the QuantiFast SYBRGreen RT-PCR Kit (Qiagen) with specific mouse oligonucleotides. The sense and antisense oligonucleotides used were, respectively, the following: 36B4, 5′-TCCAGGCTTTGGGCATCA-3′ and 5′-CTTTATCAGCTGCACATCACTCAGA-3′; pro-IL-1β, 5′-TTGACGGACCCCAAAAGATG-3′ and 5′-AGAAGGTGCTCATGTCCTCAT-3′. Each sample was run in duplicate. All results were normalized to the unaffected housekeeping 36B4 gene.
In vivo Salmonella infection. WT and TLR5KO mice were fasted 4 h and infected orally with either flagellate or aflagellate S. Typhimurium (1 × 108 colony-forming unit/mouse) and bled after 12 h for sIL-1Ra analysis. Streptomycin pretreated Salmonella-induced gastroenteritis was induced in BALB/CJ mice as previously described42 and 48 h post-infection, serum was collected for sIL-1Ra estimation.
Colon culture. Mice (WT, TLR5KO, IPAFKO, IPAF/TLR5 double knockout) were given either PBS or flagellin (10 μg/mouse in 0.2 ml PBS) i.p. and, after 1 h, colons (1 cm) were removed, cut open longitudinally, washed in HBSS and cultured in RPMI 1640 medium containing penicillin and streptomycin.15 After 24 h incubation at 37°C with 5% CO2, the supernatants were centrifuged at 4°C and used for assaying cytokines by ELISA.
Tissue MPO assay. Neutrophil influx in tissue was accessed by assaying MPO enzyme activity, a marker for neutrophils. Briefly, tissue (50 mg/ml) was thoroughly washed in PBS and homogenized in 0.5% hexadecyltrimethylammonium bromide (Sigma) in 50 mM PBS (pH 6.0), freeze-thawed three times, sonicated and centrifuged. MPO was assayed in the clear supernatant by adding 1 mg/ml of dianisidine dihydrochloride (Sigma) and 5 × 10−4% H2O2, and the change in optical density was measured at 450 nm. Human neutrophil MPO (Sigma) was used as standard. One unit of MPO activity was defined as the amount that degraded 1.0 μmol of peroxide/min at 25°C.43
ELISA. All ELISA kits are Duoset kits from R&D Systems, and assays were performed according to the manufacturer's protocol.
Statistical analysis. Significance was determined using Student's t-test (GraphPad Prism software, San Diego, CA). Differences were noted as significant *P < 0.05.
References
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Miao, E.A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575 (2006).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006).
Lightfield, K.L. et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178 (2008).
Means, T.K., Hayashi, F., Smith, K.D., Aderem, A. & Luster, A.D. The Toll-like receptor 5 stimulus bacterial flagellin induces maturation and chemokine production in human dendritic cells. J. Immunol. 170, 5165–5175 (2003).
Vijay-Kumar, M. et al. Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. Am. J. Pathol. 169, 1686–1700 (2006).
Fink, S.L. & Cookson, B.T. Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol. 9, 2562–2570 (2007).
Aksentijevich, I. et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl. J. Med. 360, 2426–2437 (2009).
Arend, W.P., Malyak, M., Guthridge, C.J. & Gabay, C. Interleukin-1 receptor antagonist: role in biology. Annu. Rev. Immunol. 16, 27–55 (1998).
Molnarfi, N., Hyka-Nouspikel, N., Gruaz, L., Dayer, J.M. & Burger, D. The production of IL-1 receptor antagonist in IFN-beta-stimulated human monocytes depends on the activation of phosphatidylinositol 3-kinase but not of STAT1. J. Immunol. 174, 2974–2980 (2005).
Hirsch, E., Irikura, V.M., Paul, S.M. & Hirsh, D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc. Natl. Acad. Sci. USA 93, 11008–11013 (1996).
Irikura, V.M., Lagraoui, M. & Hirsh, D. The epistatic interrelationships of IL-1, IL-1 receptor antagonist, and the type I IL-1 receptor. J. Immunol. 169, 393–398 (2002).
Irikura, V.M., Hirsch, E. & Hirsh, D. Effects of interleukin-1 receptor antagonist overexpression on infection by Listeria monocytogenes. Infect. Immun. 67, 1901–1909 (1999).
Vijay-Kumar, M. et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest. 117, 3909–3921 (2007).
Vijay-Kumar, M. et al. Toll-like receptor 5-deficient mice have dysregulated intestinal gene expression and nonspecific resistance to Salmonella-induced typhoid-like disease. Infect. Immun. 76, 1276–1281 (2008).
Sanders, C.J. et al. Induction of adaptive immunity by flagellin does not require robust activation of innate immunity. Eur. J. Immunol. 39, 359–371 (2009).
Vijay-Kumar, M., Carvalho, F.A., Aitken, J.D. & Gewirtz, A.T. IPAF or TLR5 is necessary and sufficient for promotion of humoral immunity by flagellin (in press).
Daig, R. et al. Human intestinal epithelial cells secrete interleukin-1 receptor antagonist and interleukin-8 but not interleukin-1 or interleukin-6. Gut 46, 350–358 (2000).
Gewirtz, A.T., Navas, T.A., Lyons, S., Godowski, P.J. & Madara, J.L. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J. Immunol. 167, 1882–1885 (2001).
Muhl, H. & Pfeilschifter, J. Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int. Immunopharmacol. 3, 1247–1255 (2003).
Paulukat, J. et al. Expression and release of IL-18 binding protein in response to IFN-gamma. J. Immunol. 167, 7038–7043 (2001).
Yu, Y. et al. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G282–G290 (2003).
Gewirtz, A.T. et al. Salmonella typhimurium induces epithelial IL-8 expression via Ca(2+)-mediated activation of the NF-kappaB pathway. J. Clin. Invest. 105, 79–92 (2000).
Uematsu, S. et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 7, 868–874 (2006).
Casini-Raggi, V. et al. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J. Immunol. 154, 2434–2440 (1995).
Sanders, C.J., Moore, D.A. III, Williams, I.R. & Gewirtz, A.T. Both radioresistant and hemopoietic cells promote innate and adaptive immune responses to flagellin. J. Immunol. 180, 7184–7192 (2008).
Fournier, B., Williams, I.R., Gewirtz, A.T. & Neish, A.S. Toll-like receptor 5-dependent regulation of inflammation in systemic Salmonella enterica Serovar typhimurium infection. Infect. Immun. 77, 4121–4129 (2009).
Schmitt, C.K. et al. Absence of all components of the flagellar export and synthesis machinery differentially alters virulence of Salmonella enterica serovar Typhimurium in models of typhoid fever, survival in macrophages, tissue culture invasiveness, and calf enterocolitis. Infect. Immun. 69, 5619–5625 (2001).
Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 87, 2095–2147 (1996).
Weber, A., Wasiliew, P. & Kracht, M. Interleukin-1beta (IL-1beta) processing pathway. Sci. Signal 3, cm2.
Arend, W.P. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 13, 323–340 (2002).
Dinarello, C.A. The role of the interleukin-1-receptor antagonist in blocking inflammation mediated by interleukin-1. N Engl. J. Med. 343, 732–734 (2000).
Arend, W.P. Interleukin-1 receptor antagonist. Adv. Immunol. 54, 167–227 (1993).
Mayer-Barber, K.D. et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J. Immunol. 184, 3326–3330.
Ivison, S.M. et al. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm. Bowel. Dis. 16, 401–409 (2010).
Vijay-Kumar, M. et al. Metabolic syndrome and altered gut microbiota in mice lacking toll-like receptor 5. Science 328, 228–231 (2010).
Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103 (2001).
Hultgren, O.H., Berglund, M., Bjursten, M. & Hultgren Hornquist, E. Serum interleukin-1 receptor antagonist is an early indicator of colitis onset in Galphai2-deficient mice. World J. Gastroenterol. 12, 621–624 (2006).
Boudeau, J., Glasser, A.L., Masseret, E., Joly, B. & Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn's disease. Infect. Immun. 67, 4499–4509 (1999).
Glasser, A.L. et al. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 69, 5529–5537 (2001).
Barthel, M. et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71, 2839–2858 (2003).
Castaneda, F.E. et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology 129, 1991–2008 (2005).
Acknowledgements
This work was supported by grants from the Crohn's and Colitis Foundation of America, and the NIH (DK061417) to AT Gewirtz. M Vijay-Kumar is a recipient of a Career Development Award from the Crohn's and Colitis Foundation of America and NIH K01 Grant DK083275-01. We also acknowledge NIH Digestive Disease Research and Development Center (DDRDC) grants to Emory University (DK064399).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Carvalho, F., Aitken, J., Gewirtz, A. et al. TLR5 activation induces secretory interleukin-1 receptor antagonist (sIL-1Ra) and reduces inflammasome-associated tissue damage. Mucosal Immunol 4, 102–111 (2011). https://doi.org/10.1038/mi.2010.57
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2010.57
This article is cited by
-
Delirium due to hip fracture is associated with activated immune-inflammatory pathways and a reduction in negative immunoregulatory mechanisms
BMC Psychiatry (2022)
-
Mobilan: a recombinant adenovirus carrying Toll-like receptor 5 self-activating cassette for cancer immunotherapy
Oncogene (2018)
-
Lipocalin 2 alleviates iron toxicity by facilitating hypoferremia of inflammation and limiting catalytic iron generation
BioMetals (2016)
-
Amino acids 89–96 of Salmonella typhimurium flagellin represent the major domain responsible for TLR5-independent adjuvanticity in the humoral immune response
Cellular & Molecular Immunology (2015)
-
Utilizing bacterial flagellins against infectious diseases and cancers
Antonie van Leeuwenhoek (2014)
