
Abstract
In healthy individuals, antigens from the gut lumen are tolerated by the mucosal immune system. However, a loss of tolerance toward the bacterial microflora probably causes inflammatory bowel disease (IBD), Crohn's disease (CD) and ulcerative colitis. The abnormal activation of the immune system in the gut of IBD patients is characterized by a cascade of cellular events orchestrated by cytokine cross talk between immune and non-immune cells. Interleukin (IL)-21, the newest member of the common γ-chain-dependent cytokine family, is a key component of the inflammatory cascade in the gut. It is highly expressed in CD and sustains the ongoing T helper type 1 (Th1)-mediated immune response. IL-21 is essential for the differentiation of Th17 cells. IL-21 is also involved in recruiting T cells to the inflamed gut and eliciting the secretion of matrix-degrading enzymes by gut fibroblasts. Overall, there is now sufficient evidence to suggest that targeting IL-21 will be of therapeutic benefit in IBD.
Similar content being viewed by others

Introduction
In healthy individuals, the gut mucosa is populated by a large number of lymphocytes in a state of balanced physiologic inflammation between the mucosal immune system and antigens of the diet and the indigenous microbiota. The mucosal immune system of the gut deals with harmless antigens by eliciting a tolerogenic immune response. In contrast, antigens produced by pathogens that colonize the intestinal tract or use the gut as a portal toward systemic infection elicit an inflammatory immune response aimed at clearing the infection. Maintaining the balance between the mucosal immune system and the antigens present in the gut lumen is pivotal for the maintenance of the intestinal immune homeostasis. A breakdown in this balance as a result of complex genetic and environmental factors is thought to underlie the pathogenesis of Crohn's disease (CD) and ulcerative colitis (UC), the two major forms of inflammatory bowel disease (IBD). IBDs are characterized by the chronic activation of the intestinal immune system resulting in gut inflammation and the downstream pathways responsible for tissue damage.
The excessive activation of the mucosal immune system in IBD is the result of a complex series of events characterized by cross talk between immune and non-immune cells via cytokines and membrane-bound receptors. The knowledge obtained from studies regarding cytokines and their role in inflammation has led to the development of novel therapeutic approaches, which are exemplified by the tumor necrosis factor-α (TNF-α) blockade in routine clinical use and by the blockade of interferon (IFN)-γ, interleukin (IL)-6, and IL-12, all being shown to be of therapeutic benefit in randomized controlled trials.1, 2, 3 However, the incomplete control of inflammation obtained using these therapies and the unresponsiveness of the majority of patients taking new biological therapies means that we still need to better define the immunologic process leading to inflammation and inflammation-related tissue damage in the gut to produce new and more effective therapies.
IL-21 is a relatively recently described cytokine, which appears to be involved at many levels in the upstream and downstream pathways of inflammation in IBD. Box 1 summarizes its biological activity. Here, we review data on the role of IL-21 in the development and maintenance of gut inflammation.
Activated Cd4+ T Cells Accumulate in the Gut in Ibd
CD4+ cells play a key role in the immune–inflammatory response leading to IBD. In both CD and UC, the inflamed mucosa shows an increased accumulation of activated CD4+ T lymphocytes. The essential role of CD4+ T cells in the development of intestinal inflammation is well demonstrated in animal models where depletion of CD4+ T cells and/or inhibition of their activation limit mucosal inflammation.4 CD4+ T cells accumulate in the gut mucosa as a result of an enhanced production of chemoattractants within the inflammatory microenvironment. However, the absolute number of CD4+ T cells present in the gut mucosa of IBD patients is also due to an enhanced rate of cell cycling in CD.5 There is a higher proliferation rate of CD4+ T cells isolated from the gut of CD patients compared to UC or normal controls. At an intracellular level, the high proliferation rate observed in CD CD4+ T cells is associated with an increased phosphorylation of Rb, which promotes the entry into the S phase of the cell cycle, and decreased phosphorylation of p53, a suppressor of S phase transit.6 In addition, T cells infiltrating the gut mucosa of IBD patients show an increased resistance to apoptosis. The mechanism of resistance is unknown, but is probably multifactorial through the activation of specific intracellular survival pathways by cytokines. In this context, blocking IL-6 has been shown to promote T-cell death, thus dampening mucosal inflammation.7 Although the direct role of IL-21 on CD4+ T-cell survival/apoptosis is unknown, signals delivered by cytokines sharing the γ-chain receptor subunit (i.e., IL-2, IL-4, IL-9, IL-13, IL-15, and IL-21) have been implicated in the maintenance and expansion of the CD4+ cell pool. Moreover, IL-21 has been shown to promote proliferation of B cells and CD8+ T cells.8
A Th1 Immune Response is a Feature of Cd
The inflammatory response seen in CD mucosa is characterized, at least in the late chronic phase, by an abundance of T helper type 1 (Th1) cells. Accordingly, lamina propria CD4+ T cells isolated from CD-affected patients produce high levels of IFN-γ. The predominant differentiation of Th1 cells fits well with the presence in diseased Crohn's mucosa of IL-12, a heterodimeric cytokine composed of p35 and p40 subunits, which drives Th1 polarization.9 IL-12, produced by activated antigen-presenting cells, targets CD4+ T cells, which express high levels of IL-12R β-chain and activate the intracellular signal transduction and activation of the signal transducers and activators of transcription (Stat)-4, a transcription factor that acts directly on the IFN-γ promoter. Moreover, IL-12 stimulation leads to T-cell expression of T-bet, a member of the family of T box transcription factors, the master controller of the Th1 lineage.10 More recently, it has also been demonstrated that IL-12 p40 can associate with a p19 subunit to make IL-23, which also promotes Th1 differentiation.11 IL-23 has been reported to be elevated in CD, but the balance of IL-12- vs. IL-23-driven Th1 differentiation is not known.12 Regardless of this, however, anti-IL-12 p40 has some clinical benefit in CD, and it neutralizes both IL-12 and IL-23.2 However, accumulating evidence has implicated IL-23 in the maintenance of Th17-mediated immune response, suggesting that this novel class of helper cells might contribute to CD pathogenesis.13
The role of IL-12 and the induction of a Th1-mediated immune response have been studied in animal models. Intrarectal administration of 2,4,6-trinitrobenzene sulfonic acid in mice causes a Th1-mediated acute colitis, which is characterized by high levels of IL-12 and IFN-γ. In this model, the neutralization of IL-12 by means of a p40-neutralizing antibody has been shown to prevent colitis.14
The Additional Role of Il-21 In Mediating Th1 Immune Response in the Gut Mucosa
The identification of the key role played by Th1 cells in colitis and of the stimuli necessary for naive T cells to polarize into Th1 cells brings into question whether other signals are required to maintain and expand Th1 responses. We have recently suggested that the type 1 cytokine, IL-21, may accomplish this function in IBDs.15
IL-21 receptor (IL-21R) was discovered 7 years ago by a genomic and expressed sequence tag sequencing approach and was identified as a novel orphan type 1 cytokine receptor.8 IL-21R shows significant structural homology with the IL-2R β-chain. IL-21 was identified by expression cloning and is structurally similar to IL-2, IL-4, IL-7, IL-9, and IL-15, which share the common γ-chain as an element of their receptor complex. Similar to other members of this family of cytokines, IL-21 interacts with a heterodimeric receptor complex formed by IL-21R and the common γ-chain. At the intracellular level, IL-21 preferentially activates Stat-1 and Stat-3 and, to a lesser extent, Stat-5A and Stat-5B. IL-21 also activates the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase intracellular pathways.
IL-21 is mainly produced by CD4+ T cells, but IL-21R is expressed by a wide range of cell types, including B and T cells, natural killer cells, myeloid cells, fibroblasts, and keratinocytes.16, 17, 18 and 19 IL-21R is expressed by both naive CD4+ and CD8+ T cells and by immature B220hi/IgMlow B cells. In both T and B cells, IL-21R is increased by ligation of both the antigen receptor and the Toll-like receptors in B cells.19 Expression of IL-21R is downregulated by IFN-α in both T and natural killer cells,20 whereas in T cells, IL-21 promotes the expression of its own receptor.21 Functionally, IL-21 augments the proliferation and cytokine profile of CD4+ and CD8 T lymphocytes.22 In B cells, IL-21 directs isotype switching and modulates both proliferation and apoptosis.23, 24
With regard to the role of IL-21 in Th1/Th2 polarization, initial studies showed that IL-21 induces the expression of Th1-related genes such as IFN-γ, T-bet, and IL-12R β-chain.22 However, in another study IL-21 was shown to inhibit IFN-γ expression in naive T cells activated under Th1 polarizing conditions without affecting the expression level of either Th1-related cytokines or T-bet.25 Therefore, the role of IL-21 in Th1 polarization remains unclear and is dependent on the specific experimental setting.
IL-21 Expression in the Gut
Analysis of IL-21 protein in biopsies of patients affected by IBD and healthy controls showed that IL-21 is overproduced in the inflamed intestine of patients with CD compared to patients with UC and healthy controls. Moreover, exogenous IL-12 further boosted IL-21 expression in explants of CD mucosa in vitro, whereas blockade of IL-21 reduced Stat-4 phosphory-lation, T-bet expression, and IFN-γ production. Collectively, these results suggest that, in CD, IL-21 may be part of a positive feedback loop that expands and maintains the ongoing Th1 cell response.15
Nevertheless, enhanced expression of IL-21 is also seen in mucosal samples from patients affected by UC, a disease that, in contrast to CD, is characterized by an atypical Th2 immune response. This suggests that the role of IL-21 in inflammation in the gut might be quite complex and that further studies are needed.
Interplay of Il-21 With other Cytokines
Accordingly, with the possible role of IL-21 in Th2-mediated immune responses, in vitro polarization of naive cells toward a Th2 phenotype enhanced IL-21 expression. Moreover, IL-21 is expressed at high levels in Balb/c mice infected with Leishmania major, which is characterized by Th2 polarization.25 IL-21 also suppresses IFN-γ expression in Th1-polarized cells without affecting T-bet expression. Moreover, in this situation, IL-21-mediated suppression of IFN-γ is not related to a downregulation of IL-12R and is mediated by a suppressive effect on Stat-4. These data suggest that in certain conditions IL-21 may dampen the expansion of a Th1 immune response by suppressing IFN-γ. In CD4+ T cells, IL-21 does not affect the expression of T-bet, but in CD8+ T cells, IL-21 suppresses the expression of eomesodermin, another member of the T box family of transcription factors, which is implicated in IFN-γ expression and in cytotoxic activity of these cells.26 Collectively, these data suggest that in certain conditions IL-21 may modulate the expression of IFN-γ in different cell types through different mechanisms, without affecting the induction of Th1 cells, as shown by the unaltered expression of T-bet.
Finally, in another model of mucosal inflammation, namely that obtained by infection of mice with Schistosoma mansoni and Nippostrongylus brasiliensis, also characterized by a Th2 immune response, IL-21-knockout mice show reduced pulmonary and hepatic granuloma formation associated with a general reduction of not only Th2 but also Th1 cytokines, suggesting that IL-21 might be involved more in the maintenance of continued T-cell activation, rather than having a unique role in T-cell polarization.27
We can then conclude that the role of IL-21 in Th1- and Th2-mediated immune responses is context dependent, enhancing both the responses rather than promoting one of the two.
Th17 Cells and Colitis
Th17 cells are a novel class of helper CD4 T cells thought to be involved in inflammatory conditions. Th17 cells, as their name implies, are characterized by the expression of IL-17, a cytokine with multiple cellular targets. Th17 cells may also express TNF-α and IL-6 but a little or no IFN-γ. Th17 cells express the orphan nuclear receptor retinoic acid-related orphan receptor-γt (RORγt), which orchestrates the differentiation of this effector cell lineage, as RORγt expression is sufficient to induce IL-17 in naive T cells.28
The coordinated activity of transforming growth factor-β (TGF-β) and IL-6 has been shown to be required to induce RORγt and IL-17 expression in naive mouse T cells.29, 30 It is noteworthy to mention that TGF-β stimulation of naive T cells has also been linked to the peripheral generation of regulatory T cells, which play a key role in the maintenance of the mucosal immune system homeostasis.31, 32 Therefore, IL-6, a cytokine highly expressed in inflammatory conditions such as IBDs, might preferentially drive the differentiation of Th17 T cells while preventing the generation of regulatory T cells.33
IL-23 has also been shown to expand the number of Th17 T cells and to augment IL-17 expression in these cells. Further, IL-23 is also highly expressed in Th17-mediated inflammatory conditions.34, 35
Studies in mice indicate the importance of Th17 T cells in the generation and maintenance of mucosal inflammation and inflammation-related tissue damage. IL-10-knockout mice develop severe colitis. IL-10/IL-12p35 double knockout mice show an equal susceptibility to colitis, whereas IL-10/IL-23p19 double knockout mice do not develop colitis,36 thus indicating that the IL-23/Th17, but not the IL-12/Th1, axis of the immune system is involved in the development of colitis induced by IL-10 deficiency. In an adoptive transfer model of colitis, administration of exogenous IL-23 exacerbated gut inflammation. Likewise, neutralization of p19IL-23 protected C3H/HeJBir mice from colitis.37 p19-deficient Rag−/− mice were also protected from colitis induced upon adoptive transfer of naive T cells, whereas p40- or p35-knockout mice were susceptible. Consistent with this, p19-knockout mice were also highly susceptible to 2,4,6-trinitrobenzene sulfonic acid colitis in comparison to both p35-knockout and wild-type mice.38 Overall, these data indicate that Th17 and Th1 T cells mediate different types of colitis, as underscored by the different outcomes obtained by IL-23/IL-12 blockade in different mice models. However, it is not possible at the moment to rule out the possibility that Th17 and Th1 immune responses might coexist, representing different stages of the same mucosal inflammatory process.
High levels of IL-23 and IL-17 have been observed in the colon of CD-affected patients.39, 40 In addition, mutations of the gene encoding for an IL-23R subunit, located on the chromosome 1p31, have been linked to CD and UC susceptibility, thus linking the IL-23/Th17 axis to IBD.41
IL-21 and Th17
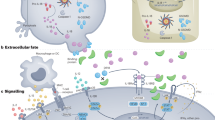
Recently, several groups have independently shown that in addition to IL-6 and TGF-β, IL-21 is required for the de novo induction of Th17 cells42, 43, 44, 45, 46 in mice (Figure 1). The resistance to Th17-mediated inflammation in experimental autoimmune encephalomyelitis, seen in IL-6-deficient mice and linked to the absence of Th17 cells, is abrogated by the in vivo depletion of regulatory T cells, thus suggesting that cytokines other than IL-6 are involved in Th17 differentiation, in the absence of regulatory T cells. Analysis of Th17 differentiation in different in vitro models has shown that the endogenous expression of IL-21 driven by IL-6 stimulation is required for the generation of Th17 cells in the presence of TGF-β. However, whether autocrine IL-21 expression is required for the IL-6-mediated induction of Th17 cells or whether IL-21 helps amplify this process needs to be better defined. Indeed, the expression of the IL-23R, a cytokine involved in the maintenance/expansion of Th17 cells, was enhanced by IL-21, but not IL-6, whereas IL-6 was still able to induce a certain IL-17 expression also in the absence of the IL-21R,42 thus suggesting that IL-21 might be involved in the amplification of Th17-mediated immune response. Interestingly, IL-21 is expressed by Th17 cells, thus suggesting that it may sustain Th17-mediated inflammation in a paracrine manner.

IL-6-mediated IL-21 autocrine expression promotes Th17 cell differentiation and blocks inducible Treg (iTreg) induction. IFN-γ, interferon-γ; IL, interleukin; TGF-β, transforming growth factor-β; Th, T helper cell.
As IL-21 is highly expressed in both CD and UC, which are characterized by a dominant Th1- and Th2-like immune response, respectively, it is tempting to speculate that in IBD an exaggerated Th17 inflammatory response might be a common event. There is evidence that mucosal T cells from Crohn's patients express abundant IL-17, but this has not been studied systematically in UC. However, it is worth noting that in humans the role of IL-6 and TGF-β in Th17 cell generation has been questioned, raising the possibility that the role of IL-21 in man may also differ from what is observed in mice.
Il-21 Targets Intestinal Epithelial Cells and Enhances T-Cell Chemotaxis
Gut epithelial cells play an important role in immune response, and their capacity to respond to bacteria, present antigens, and secrete cytokines, which control survival and activity of mucosal lymphocytes, makes them an integral part of the innate immunity. Moreover, intestinal epithelial cells sustain inflammation by expressing high levels of chemoattractants, which cause activated lymphocytes to accumulate in the gut mucosa. Vice versa, activated lymphocytes influence epithelial cell activity through cytokine expression. Therefore, the interaction of lymphocytes and gut epithelial cells represents an important aspect of the cross talk between innate and adoptive immunity.
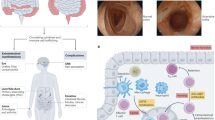
Intestinal epithelial cells constitutively express the IL-21R complex. In addition, IL-21R expression is further enhanced in the inflamed tissue of UC and CD compared to uninflamed, normal controls, suggesting that IL-21 might directly target intestinal epithelial cells during inflammation. In vitro experiments using epithelial cell lines have shown that IL-21 increases secretion of the macrophage inflammatory protein (MIP)-3α (CCL20).47 Accordingly, in both CD and UC mucosa, where IL-21 is expressed at high levels, MIP-3α is upregulated, which probably contributes to the recruitment of gut-homing α4-β7-expressing T cells (Figure 2). Indeed, MIP-3α accounts for most of the T-cell chemoattractant activity of intestinal epithelial cell cultures. Recent data show that T cells sharing features of both Th1 and Th17 cells have isolated from the lamina propria of CD-affected patients. These cells are characterized by the expression of CC chemokine receptor 6, the MIP-3α receptor, thus suggesting that MIP-3α might favor the accumulation of these cells into the gut lamina propria during chronic inflammation.48

The picture illustrates some of the biological functions of IL-21 in the gut. IL-21 enhances the production of IFN-γ by Th1 cells. IL-21 also stimulates fibroblasts to synthesize MMPs, a family of proteases that cleave components of the extracellular matrix, thereby promoting tissue damage. Additionally, IL-21 enhances the production of MIP-3α, a T-cell chemoattractant, in epithelial cells. IFN-γ, interferon-γ; IL-21, interleukin-21; MIP-3α, macrophage inflammatory protein-3α; MMP, matrix metalloproteinase; Th1, T helper type 1.
Il-21 Contributes to Gut Damage by Enhancing the Secretion of Metalloproteases
In CD and UC, inflammatory flares cause erosions and ulceration frequently involving large areas of the intestinal mucosa. However, after years of active disease, the physiologic response to the inflammation-related damage results in the subversion of the original organ structure characterized by an abnormal deposition of collagen in CD. The main players in this process are lamina propria myofibroblasts and fibroblasts, which produce collagen, profibrotic factors and, in inflammatory conditions, large amounts of matrix metalloproteinases (MMPs). MMPs are a family of neutral endopeptidases that can cleave multiple components of the extracellular matrix. These enzymes are mostly secreted as proenzymes and activated extracellularly. Their activity is tightly controlled by specific inhibitors of metalloproteinases (tissue inhibitors of metalloproteinases). An imbalance between MMPs and their inhibitors results during inflammation, implicating that inflammatory mediators may contribute to the tissue damage observed in chronic inflammation.
We have recently demonstrated that IL-21R is expressed by gut fibroblasts and that IL-21 stimulation results in the enhanced secretion of MMP-1, MMP-2, MMP-3, and MMP-9 but not tissue inhibitors of metalloproteinases.49 Neutralization of IL-21 markedly reduces the amounts of MMPs released when fibroblasts are cultured with CD lamina propria mononuclear cell supernatants, confirming the role of IL-21 as mediator of the MMP-mediated tissue damage (Figure 2). Interestingly, IL-21 does not affect the expression of MMPs at the transcriptional or post-transcriptional level, rather it acts by causing the release of intracellular stores of MMPs.
TNF-α cooperates with IL-21 in inducing MMP secretion by fibroblasts. A possible mechanism to explain this cooperative effect is that TNF-α, similar to the other proinflammatory cytokine IL-1β, increases expression of IL-21R in these cells, thus enhancing IL-21 signaling.
Although the role of IL-21 in gut fibrosis has not been investigated in animal models of colitis, data produced in a liver fibrosis model after infection with S. mansoni indicate that mice lacking the IL-21R develop significantly less fibrosis compared to wild-type mice.27 However, as IL-21 is implicated in the generation of Th17 cells and high levels of IL-17 have been observed in this model, it is not possible to rule out the possibility that the difference in fibrosis observed in IL-21R-deficient mice is secondary to a less severe Th17 immune response.
Concluding Remarks
Mucosal inflammation is the result of the cross talk among different cell lineages. This cross talk is controlled by cytokines and their interaction with specific receptors expressed on the surface of target cells. As a consequence, many phenomena observed during mucosal inflammation result from the response to cytokines of specific cell types. In IBD chronic inflammation of the gut is orchestrated by the coordinated release of cytokines, which initiate and sustain the cellular events responsible for initiation and maintenance of the inflammatory process. For instance, in CD the massive release of IL-12 by antigen-presenting cells creates an environment that favors the differentiation of naive T cells into Th1 cells. In turn, Th1 cells produce IFN-γ, thus amplifying the Th1-mediated inflammation characterized by the massive release of TNF-α, an important mediator of inflammation-related tissue damage.
IL-21, a T-cell-derived cytokine, also exerts multiple effects on the inflammatory process. Here, we have summarized data showing that IL-21 is involved in the de novo differentiation of Th17 cells, a novel class of T helper cells that plays a pivotal role in many inflammatory diseases such as rheumatoid arthritis, multiple sclerosis, and psoriasis. Moreover, IL-21 contributes to the accumulation of T cells in the lamina propria of IBD patients by enhancing the chemoattractant MIP-3α. Finally, by modulating the expression of metalloproteases, IL-21 is directly implicated in the inflammation-related tissue damage and architectural subversion observed in the gut of IBD patients affected by long-standing inflammation.
The relevant therapeutic benefit gained by using monoclonal antibodies against TNF-α in IBD therapy has pushed research to find novel molecular targets to dampen chronic immune responses. For instance, a novel anti-p40 antibody that targets the common subunit of both IL-12 and IL-23, thus blocking both Th1 and Th2 immune responses, has shown promising results in human trials. However, in Th17-mediated diseases, the selective inhibition Th17 cells, while keeping other immune responses (i.e., Th1 and Th2) unaffected, might be safer inasmuch as it could avoid excessive immunosuppression. The effect of IL-21 in mucosal inflammation and its role in Th17 cell generation makes this cytokine an interesting target for therapy, thus representing a possible alternative to anti-TNF-α and anti-p40 monoclonal antibodies in IBD therapy.
Disclosure
The authors declared no conflict of interest.
References
van Assche, G. Emerging drugs to treat Crohn's disease. Expert Opin. Emerg. Drugs 12, 49–59 (2007).
Mannon, P.J. et al. Anti-interleukin-12 antibody for active Crohn's disease. N. Engl. J. Med. 351, 2069–2079 (2004).
Ito, H. et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn's disease. Gastroenterology 126, 989–996 (2004).
Takahashi, I., Kiyono, H. & Hamada, S. CD4+ T-cell population mediates development of inflammatory bowel disease in T-cell receptor alpha chain-deficient mice. Gastroenterology 112, 1876–1886 (1997).
Sturm, A. et al. Divergent cell cycle kinetics underlie the distinct functional capacity of mucosal T cells in Crohn's disease and ulcerative colitis. Gut 53, 1624–1631 (2004).
Ina, K. et al. Resistance of Crohn's disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J. Immunol. 163, 1081–1090 (1999).
Atreya, R. et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat. Med. 6, 583–588 (2000).
Parrish-Novak, J. et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 408, 57–63 (2000).
Monteleone, G. et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology 112, 1169–1178 (1997).
Neurath, M.F. et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J. Exp. Med. 195, 1129–1143 (2002).
Oppmann, B. et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 (2000).
Schmidt, C. et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm. Bowel Dis. 11, 16–23 (2005).
Iwakura, Y. & Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Invest. 116, 1218–1222 (2006).
Neurath, M.F., Fuss, I., Kelsall, B.L., Stuber, E. & Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 (1995).
Monteleone, G. et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn's disease. Gastroenterology 128, 687–694 (2005).
Brandt, K., Bulfone-Paus, S., Foster, D.C. & Ruckert, R. Interleukin-21 inhibits dendritic cell activation and maturation. Blood 102, 4090–4098 (2003).
Brandt, K., Bulfone-Paus, S., Jenckel, A., Foster, D.C., Paus, R. & Ruckert, R. Interleukin-21 inhibits dendritic cell-mediated T cell activation and induction of contact hypersensitivity in vivo. J. Invest. Dermatol. 121, 1379–1382 (2003).
Distler, J.H. et al. Expression of interleukin-21 receptor in epidermis from patients with systemic sclerosis. Arthritis Rheum. 52, 856–864 (2005).
Jin, H., Carrio, R., Yu, A. & Malek, T.R. Distinct activation signals determine whether IL-21 induces B cell costimulation, growth arrest, or Bim-dependent apoptosis. J. Immunol. 173, 657–665 (2004).
Strengell, M., Julkunen, I. & Matikainen, S. IFN-alpha regulates IL-21 and IL-21R expression in human NK and T cells. J. Leukoc. Biol. 76, 416–422 (2004).
Zeng, R. et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 201, 139–148 (2005).
Strengell, M., Sareneva, T., Foster, D., Julkunen, I. & Matikainen, S. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J. Immunol. 169, 3600–3605 (2002).
Ozaki, K. et al. A critical role for IL-21 in regulating immunoglobulin production. Science 298, 1630–1634 (2002).
Ozaki, K. et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 173, 5361–5371 (2004).
Wurster, A.L. et al. Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J. Exp. Med. 196, 969–977 (2002).
Suto, A., Wurster, A.L., Reiner, S.L. & Grusby, M.J. IL-21 inhibits IFN-gamma production in developing Th1 cells through the repression of Eomesodermin expression. J. Immunol. 177, 3721–3727 (2006).
Pesce, J. et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J. Clin. Invest. 116, 2044–2055 (2006).
Ivanov, I.I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Mangan, P.R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234 (2006).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
Chen, W. et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Fantini, M.C., Becker, C., Monteleone, G., Pallone, F., Galle, P.R. & Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 172, 5149–5153 (2004).
Dominitzki, S. et al. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J. Immunol. 179, 2041–2045 (2007).
Langrish, C.L. et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005).
Harrington, L.E. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 (2005).
Yen, D. et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316 (2006).
Elson, C.O. et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 (2007).
Becker, C. et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 177, 2760–2764 (2006).
Fuss, I.J. et al. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 12, 9–15 (2006).
Fujino, S. et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003).
Duerr, R.H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006).
Zhou, L. et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8, 967–974 (2007).
Korn, T. et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448, 484–487 (2007).
Nurieva, R. et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448, 480–483 (2007).
Wei, L., Laurence, A., Elias, K.M. & O’Shea, J.J. IL-21 is produced by TH17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 282, 34605–34610 (2007).
Fantini, M.C. et al. IL-21 regulates experimental colitis by modulating the balance between T(reg) and Th17 cells. Eur. J. Immunol. 37, 3155–3163 (2007).
Caruso, R. et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology 132, 166–175 (2007).
Annunziato, F. et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861 (2007).
Monteleone, G. et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 55, 1774–1780 (2006).
Acknowledgements
This work was supported by Eli and Edythe L Broad Foundation; the “Foundation Umberto Di Mario,” Rome, Italy; and Giuliani SpA, Milan, Italy.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Fantini, M., Monteleone, G. & MacDonald, T. IL-21 comes of age as a regulator of effector T cells in the gut. Mucosal Immunol 1, 110–115 (2008). https://doi.org/10.1038/mi.2007.17
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2007.17
This article is cited by
-
Functional roles of cytokines in infectious disease associated colorectal carcinogenesis
Molecular Biology Reports (2022)
-
Role of interleukin-21 in HBV infection: friend or foe?
Cellular & Molecular Immunology (2015)
-
Interleukin-21: a double-edged sword with therapeutic potential
Nature Reviews Drug Discovery (2014)
-
Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease
Inflammation Research (2014)
