
Abstract
The remarkable narrow-band emission of trivalent lanthanide-doped phosphors excited by the vacuum ultraviolet (VUV) radiation lines of Xe atoms/Xe2 molecules at 147/172 nm are extensively investigated in the development of plasma display panels and Hg-free fluorescent lamps, which are frequently used in our daily lives. Numerous solid materials, particularly Tb3+-doped oxides, such as silicates, phosphates and borates, are efficient green/blue sources with color-tunable properties. The excitation wavelength and rare earth concentration are usually varied to optimize efficiency and the luminescent properties. However, some underlying mechanisms for the shift in the emission colors remain unclear. The present study shows that a UV/VUV switch systematically controls the change in the phosphor (Ba3Si6O12N2:Tb) photoluminescence from green to blue, resulting in a green emission when the system is excited with UV radiation. However, a blue color is observed when the radiation wavelength shifts to the VUV region. Thus, a configurational coordinate model is proposed for the color-reversal effect. In this model, the dominant radiative decay results in a green emission under low-energy UV excitation from the 5D4 state of the f–f inner-shell transition in the Tb system. However, under high-energy VUV excitation, the state switches into the 5D3 state, which exhibits a blue emission. This mechanism is expected to be generally applicable to Tb-doped phosphors and useful in adjusting the optical properties against well-known cross-relaxation processes by varying the ratio of the green/blue contributions.
Similar content being viewed by others

Introduction
Inorganic-material-based phosphors have been extensively investigated for their applications in electronic illustrations, such as backlighting sources of liquid–crystal displays, plasma display panels and white light-emitting diodes (WLEDs)1, 2. In particular, phosphors are important components of these displays, which have chemical durability and efficient luminescent properties. Compared with the traditional incandescent lamp and mercury-vapor lamp, WLED has attracted considerable attention because of its highly applicable value for our daily lives and wide feasibility for use in commercial products3, 4. The most common WLED strategy is to combine blue InGaN chips and Y3Al5O12:Ce3+ (YAG:Ce) phosphor in addition to employing three light-emitting diode (LED) chips in red, green and blue, which partially converts the original blue radiation into the complementary yellow color, yielding cool white light. This cool white light, which is based on using a single phosphor, is suitable for everyday applications only if the poor color rendition index (CRI) and high-correlated color temperature are bearable5, 6. The requirement for high CRI cannot be satisfied using this approach because the insufficient spectral components cannot entirely cover the visible region. Recently, a novel LED device, in which white light is produced by an ultraviolet (UV) chip with red, green and blue phosphors, can achieve a high CRI of up to 907, 8. To pursue the above purposes, new phosphors adopted for UV excitation must be developed.
Oxonitridosilicates, which are formally derived from typical oxosilicates by partially substituting oxygen with nitrogen to form rigid Si(O,N)4 tetrahedra, have excellent chemical, physical, mechanical and thermal stabilities for application in WLEDs9, 10, 11. With additional structural possibilities, nitrogen can be a triple or even quadruple connecting atom in the tetrahedral network. Therefore, the diversity of oxonitridosilicates is superior to that of oxosilicates because only terminal- and simple-bridging oxygen are available in oxosilicates12.
A system of Ba3Si6Ox=6, 9, 12, 15Ny=6, 4, 2, 0:M (M=Eu2+, Ce3+) has been reported because of its high luminescent properties and easy synthesis13. In 2009, Mikami et al14 first reported the green oxynitride phosphor Ba3Si6O12N2:Eu. The structural characterization and luminescent properties of Ba3−xSrxSi6O12N2 (with x=0.4 and 1) were studied by Braun et al15. Tang et al16 utilized first-principle calculations to confirm the high-luminescence intensity of the Ba3Si6O12N2:Eu2+ phosphor, which has a direct band gap and a low-energy band dispersion. To date, terbium (Tb)-doped Ba3Si6O12N2 has not been reported. In contrast, Tb-activated phosphors are widely studied as green or blue luminescent candidates in the development of UV/vacuum UV (VUV)-excited applications17, 18, 19, 20, 21, 22, 23, 24, 25. The relative intensities of 5D3/5D4 emissions strongly depend on the Tb concentration through the cross-relaxation process, which result in a color change from blue to green26. However, studies on the systematic photoluminescence excitations (PLEs) from UV to VUV radiation are rare, and related studies are also limited. Therefore, a Ba3Si6O12N2 material with constant 3.7% Tb doping at the Ba site was synthesized and characterized in the current study. The principal relationship between the different excitation energies and the luminescence mechanism was investigated.
Materials and methods
Synthesis of materials
Oxynitride Ba3Si6O12N2:Tb was synthesized from stoichiometric mixtures of high-purity BaCO3 (J. T. Baker, 99.9%), α-Si3N4 (Ube Industries, grade SN-E10, α/(α+β)>95% by weight), SiO2 (Aldrich, 99.995%) and Tb4O7 (Aldrich, 99.9%). The ground mixtures were placed in boron nitride crucibles and were then sintered using a graphite heater in a gas-pressure sintering furnace (FVPHP-R-5, FRET-25, Fujidempa Kogyo Co. Ltd.). The temperature was increased to 500 °C (heating rate: 5 °C min−1) under a vacuum of 10−2 Pa. Nitrogen gas (99.999% purity) was then introduced at a pressure of 0.92 MPa. The temperature was subsequently increased to 1375 °C and held for 1 h. The samples were cooled to room temperature (cooling rate: 5 °C min−1) and then powdered for subsequent analyses.
Characterization methods
Synchrotron X-ray diffraction patterns with wavelength of λ=0.774907 Å were recorded using a Debye–Scherrer camera installed at the BL01C2 beamline of the National Synchrotron Radiation Research Center, Taiwan. X-ray Rietveld profile refinements of the structural models and texture analysis were performed using the General Structure Analysis System software27. 29Si solid-state nuclear magnetic resonance spectrum was recorded on a wide-bore 14.1 Tesla Bruker Avance III NMR spectrometer (Germany), equipped with a 4-mm double-resonance magic-angle-spinning probe. The Larmor frequency for 29Si was 119.24 MHz. High-resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED) images were obtained via a JEOL JEM-2011 microscope (USA) operated at 200 kV. Synchrotron VUV photoluminescence (PL) and PLE spectra were obtained using the same synchrotron source at the BL03A beamline. The excitation spectra were recorded by scanning a 6 m cylindrical grating monochromator with a grating of 450 grooves mm−1 over a wavelength range of 100–350 nm. A CaF2 plate served as a filter to remove the high-order light from the synchrotron. The emission from the phosphor was analyzed with a 0.32 m monochromator and then detected in a photon-counting mode. PL and PLE spectra were collected using a FluoroMax-3 spectrophotometer (USA) equipped with a 150 W Xe lamp and a Hamamatsu R928 photo-multiplier tube (Japan).
Results and discussion
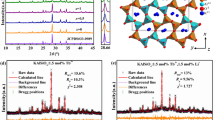
The X-ray Rietveld refinement of Ba2.89Si6O12N2:Tb0.11 (BSON:Tb) is shown in Figure 1a, including the observed, calculated and difference profiles, and the relative Bragg reflection markers. Supplementary Table S1 presents the crystallographic data, which are consistent with the lattice constants, reflection conditions and cell parameters of a previous study28. The results indicated that the compound is in pure phase, the data are reliable and the powder sample is crystallized into a trigonal structure with a  (no. 147) space group. In the inset of Figure 1a, the peak at −60 to −90 ppm indicates that Si4+ cations in the structural lattices are coordinated by oxide and nitride to form the [SiO3N] tetrahedra. The other peak at −90 to −120 ppm is ascribed to SiO2 on the surface of the material29. The results demonstrated that the structure of BSON:Tb is successfully synthesized through gas pressure sintering. HRTEM image reveals an irregular arrangement of BSON:Tb (Figure 1b), whereas the selected area electron diffraction image exhibits a specific regular shaped-pattern spots corresponding to the [001] zone axis of the trigonal structure (Figure 1c). According to the three planes {010}, {100} and {1–10} of Figure 1c, the crystal-lattice spacing is 6.482 Å, which is consistent with the pure BSON result (6.468 Å). Figure 1d shows the crystal structure of 2 × 2 × 3 unit cells, viewed along the [010] direction. The BSON structure consists of barium and SiO3N-tetrahedral layers. The distance is 6.5 Å in these layers, as shown in Figure 1e. The shared N atom is linked with the three SiO3 (see the green circle area of Figure 1f), and many [N(SiO3)3] are connected by oxygen atoms into the mesh [SiO3N] tetrahedral layers. The 6er- and 4er-rings are built by vertex-sharing SiO3N tetrahedra (Figure 1h) as the fundamental building units (see the blue dotted lines of Figure 1f). The Ba ions are located at two independent crystallographic sites. One Ba ion is surrounded by six oxygen ions in site 1, whereas the other Ba ion (in site 2) is also coordinated by six oxygen ions, but has an additional capped nitrogen ion. The coordination of these two Ba sites results in the formation of a slightly distorted octahedral structure, as shown in Figure 1g. Therefore, the Tb activators occupy these sites by replacing the Ba ions. The coordination symmetry of the surrounding anions is significant to the degeneracy of the activator 5d level in the 4f→5d transition shown below because the Tb activators are not in a perfect octahedral crystal-field environment. As heterovalent and homovalent substitutions, the PLE and PL spectra of BSON:Ce and BSON:Ce, Li are very similar, except in terms of intensity, as shown in Supplementary Fig. S1. The lattice structure retains electric neutrality through vacancies, defects and anions (O2− and N3−), although Tb3+ activators are introduced into the Ba2+ sites30, 31. Based on the structure refinement, the crystallographic data (Supplementary Table S1) demonstrated that BSON:Tb can maintain a perfect structure without charge compensation.
(no. 147) space group. In the inset of Figure 1a, the peak at −60 to −90 ppm indicates that Si4+ cations in the structural lattices are coordinated by oxide and nitride to form the [SiO3N] tetrahedra. The other peak at −90 to −120 ppm is ascribed to SiO2 on the surface of the material29. The results demonstrated that the structure of BSON:Tb is successfully synthesized through gas pressure sintering. HRTEM image reveals an irregular arrangement of BSON:Tb (Figure 1b), whereas the selected area electron diffraction image exhibits a specific regular shaped-pattern spots corresponding to the [001] zone axis of the trigonal structure (Figure 1c). According to the three planes {010}, {100} and {1–10} of Figure 1c, the crystal-lattice spacing is 6.482 Å, which is consistent with the pure BSON result (6.468 Å). Figure 1d shows the crystal structure of 2 × 2 × 3 unit cells, viewed along the [010] direction. The BSON structure consists of barium and SiO3N-tetrahedral layers. The distance is 6.5 Å in these layers, as shown in Figure 1e. The shared N atom is linked with the three SiO3 (see the green circle area of Figure 1f), and many [N(SiO3)3] are connected by oxygen atoms into the mesh [SiO3N] tetrahedral layers. The 6er- and 4er-rings are built by vertex-sharing SiO3N tetrahedra (Figure 1h) as the fundamental building units (see the blue dotted lines of Figure 1f). The Ba ions are located at two independent crystallographic sites. One Ba ion is surrounded by six oxygen ions in site 1, whereas the other Ba ion (in site 2) is also coordinated by six oxygen ions, but has an additional capped nitrogen ion. The coordination of these two Ba sites results in the formation of a slightly distorted octahedral structure, as shown in Figure 1g. Therefore, the Tb activators occupy these sites by replacing the Ba ions. The coordination symmetry of the surrounding anions is significant to the degeneracy of the activator 5d level in the 4f→5d transition shown below because the Tb activators are not in a perfect octahedral crystal-field environment. As heterovalent and homovalent substitutions, the PLE and PL spectra of BSON:Ce and BSON:Ce, Li are very similar, except in terms of intensity, as shown in Supplementary Fig. S1. The lattice structure retains electric neutrality through vacancies, defects and anions (O2− and N3−), although Tb3+ activators are introduced into the Ba2+ sites30, 31. Based on the structure refinement, the crystallographic data (Supplementary Table S1) demonstrated that BSON:Tb can maintain a perfect structure without charge compensation.

Structure determination and description. (a) X-ray Rietveld refinements of the Ba2.89Si6O12N2:Tb0.11 material. The observed (crosses), calculated (solid line) and difference profiles (bottom) are shown together with the Bragg markers. 29Si solid-state NMR spectrum of the BSON:Tb compound (inset). (b) HRTEM image of the BSON:Tb phosphor. (c) SAED pattern along the [001] zone axis. (d–f) Crystal structure of the BSON material along various directions. (g) Coordination model of two Ba sites that form a slightly distorted octahedron. (h) Si atoms are hidden in the tetrahedral inside. (Ba1: purple, Ba2: dark red, Si: green, O: red, and N: blue.).
The emission spectrum of the Tb activator normally presents two typical sets of intense line systems from the 5D4→7FJ (J=3–6, 620–465 nm) and 5D3→7FJ (J=3–6; 465–375 nm) transitions, which result in green and blue emissions, respectively. The dominant set usually corresponds to 5D4→7FJ transitions (green set) in most cases32, 33, 34, whereas the 5D3→7FJ transitions (blue set) are difficult to obtain as a primary emission because of the depopulation of the 5D3 state. This phenomenon can be elucidated by the direct feeding of the excited energy from the 5d level to the 5D4 state35. The cross-relaxation process between neighboring Tb activators also results in considerable quenching from the 5D3 to the 5D4 state, as normally expected in many cases with high-Tb-dopant concentrations26. These two mechanisms are responsible for the predominant green emissions of numerous Tb-doped phosphors. In this study, an unprecedented effect was further investigated by performing a color reversal between the green and blue sets through the control of the relative contributions of both colors in a Tb-doped phosphor under synchrotron radiation excitation at different wavelengths. The experiment had two requirements: (1) a moderate Tb concentration to ensure the initial appearance of a green emission and (2) changeable excitation energy from the UV to VUV range.
A series of PL spectra of the Ba2.89Si6O12N2:Tb0.11 phosphor is shown in Figure 2a. The phosphor was excited under several specific synchrotron radiation wavelengths: 254, 234, 211 and 147 nm (according to the excitation peaks in the PLE spectra). The characteristic fine structure in the PL spectra is caused by the splitting of the 2S+1LJ states as a result of Russell–Saunders coupling. Each specific 5DJ=3, 4 to 7FJ=3–6 transition is labeled in the inset, and the corresponding energy levels are plotted in Supplementary Fig. S2. The most intense transitions in the green and blue transition sets are 5D4→7F5 and 5D3→7F6, respectively. By contrast, PLE spectra monitored at 542 (5D4→7F5), 478 (5D4→7F6), 435 (5D3→7F4) and 412 nm (5D3→7F5) wavelengths are shown in Figure 2b. At a broad band (200–280 nm) region, three distinct peaks at 211 [4f8(7F6)→4f75d1], 234 [4f8(7F5)→4f75d1] and 254 nm [4f8(7F0,7F1)→4f75d1] in the PLE spectra are attributed to the transition from the 7F6 ground state to the 7DJ excited state, which corresponds to the 4f8 to 4f75d1 levels. At these levels, the transition belongs to the spin-allowed 4f→5d transition. The 4f→5d transition of Tb3+ in Ba3Si6O12N2 can be predicted by Dorenbos’ expression36, 37, 38, 39:

Synchrotron photoluminescence of the Ba2.89Si6O12N2:Tb0.11 material. (a) Emission spectra from synchrotron radiation excited by different wavelengths and (b) excitation spectra monitored at 5D3 and 5D4 transition sets at room temperature. The arrows illustrate the excited or monitored wavelengths from specific positions of the excitation or emission spectra, respectively.


where D(Ln, A) is the crystal-field depression of the 4fn−15d levels of a lanthanide ion (Ln3+) in compound A relative to the energies in the free ion, E(Ln, free) is the energy of the first f–d transition of Ln3+ as a free ion (gaseous), E(Ln, A) is the f–d energy difference of Ln3+-doped compound A with D(Ln, A) and ΔELn,Ce is defined as the difference in f–d energy of Ln3+ with that of the first electric dipole-allowed transition of Ce3+. The effect of the crystal field and the covalence of the host lattice on the red shift of the 5d levels are approximately equal for all rare earth ions. Thus, the depression of D(Ce3+, Ba3Si6O12N2) can be used to predict the 5d energies of other lanthanides. The lowest 4f–5d excitation transition of Ce3+, E(Ce3+, Ba3Si6O12N2), was determined as 338 nm (29 586 cm−1), as shown in Supplementary Fig. S1. The 5d level of the free Ce3+ ion was reported as 49 340 cm−1 34. Therefore, D(Ce3+, Ba3Si6O12N2) is ~19 754 cm−1 according to Equation (1). ΔETb,Ce is reported as 13 200 cm−1 34. The lowest 4f–5d transition energy E(Tb3+, Ba3Si6O12N2) can be predicted by Equation (2) and was determined as 42 786 cm−1 (233 nm), which was consistent with the experimental data (234 nm). The calculation scheme is shown in Supplementary Fig. S3.
Another broad band (130–200 nm) monitored at 435/412 nm (5D3→7F4,5; blue sets) belongs to a mixed band composed of the host-absorption band (HAB) and the O2p/N2p to Tb4f charge-transfer band transitions. Furthermore, the host-absorption band from the undoped Ba3Si6O12N2 sample is at ~180 nm. This value agrees well with the calculated band gap of 6.9 eV using density functional theory calculation16, 28. The detailed discussion and experimental results are depicted in the supporting information and Supplementary Fig. S4. The charge-transfer band position of Tb3+ in Ba3Si6O12N2 can be predicted by Jørgensen’s expression40:

where χopt(X) is the optical electronegativity of the ligand ion (similar to Pauling’s electronegativity), and χuncorr(M) can be calculated using Su’s expression41:

where E0Tb(Tb3+→Tb2+) was reported as −3.7 eV41. Therefore, χuncorr(Tb) can be predicted to be 0.95. The ECT values were predicted as 64 500 and 52 500 cm−1, which correspond to 155 and 190 nm, respectively. χopt(O) and χopt(N) are ~3.1 and ∼2.7, respectively.
A possible mechanism for the overall effects of UV/VUV-pumped Tb-doped phosphor is proposed and depicted by a configurational coordinate model shown in Figure 3a. A moderate Tb-activator concentration is used in the Ba2.89Si6O12N2:Tb0.11 phosphor (Tb occupancy of ~3.7 atom%) to suppress the high cross-relaxation probability (i.e., to retain a certain number of excited electrons at the 5D3 state). Radiative decay partly occurs from the 5D3 to the 7FJ=3–6 states. Nevertheless, the green set remains the leading transition because the direct feeding from the 5d level to the 5D4 state [path (I) in Figure 3a] remains the dominant process (shown in 254 nm of the excited PL spectrum). The green transition gradually declines with increasing excited radiation energy from 254 to 147 nm, and the blue transition concurrently grows toward the opposite direction. Increased radiation energy from 254 to 211 nm leads to enhanced probability of the excited electrons to move down to the 5D3 state by crossing the intersection point between the 5d level and the 5D3 state [path (II) in Figure 3a].

Mechanism. (a) Configurational coordinate model of the color-reversal effect. The broken arrow from VB to CB represents a color switch that is tunable and reversible between green and blue emissions. (b) Ratio of the blue set to the green set, ΣI(5D3)/I(5D4), under 254 nm UV to 147 nm VUV excitation. The ratio of the entirely integrated emission bands of 5D3→7FJ=3–6 to that of 5D4→7FJ=3–6.
Surprisingly, the blue set rapidly becomes the primary emission when the excitation energy reaches 147 nm in the VUV range. This process can be attributed to a mechanism by which the electrons of the host lattice in the valence band (VB) are excited to the conduction band (CB) by high-energy radiation (147 nm) and then relax to the charge-transfer state (CTS) [path (III) in Figure 3a]42. These electrons transfer to the 5D3 state by crossing the CTS-5D3 intersection point [path (IV) in Figure 3a] and eventually return to the ground state by emitting blue light. Therefore, the determining step, VB→CB, in the foregoing route (VB→CB→CTS→5D3 state→emitting blue light) is considered the possible driving force for the color-reversal effect. The band gap between VB and CB (host-lattice absorption) was evaluated at 6.9 eV. This value indicates that the route can be switched on or off (i.e., as a color-tunable switch) by controlling the excited wavelength at approximately this energy.
Figure 3b plots the ratio of the blue set to the green set, ΣI(5D3)/I(5D4) (i.e., the ratio of the entirely integrated emission bands of 5D3→7FJ=3–6 to that of 5D4→7FJ=3–6) according to the emission spectra of the BSON:Tb excited by different wavelengths (254, 234, 211, 190, 170 and 147 nm), as shown in Supplementary Fig. S5. Under 211–254-nm-UV excitation, this ratio is ~0.5. However, the ratio increases rapidly to 4.6 under 147-nm-VUV excitation, indicating that additional emission in the 5D3→7FJ=3–6 process can be estimated when the excitation energy exceeds 6.5 eV. This extra energy can be attributed to the electrons donated from the host VB because the onset energy for this curve is at ~6.5 eV. This value closely matches the band gap of the host lattice, suggesting that the excited electrons of the 5D3 state radiative decay are from both the 5d level of the Tb activator and the host-lattice CB. This dual accumulation leads to an exponential growth in the ΣI(5D3)/I(5D4) ratio, and the systematic color reversal between blue and green sets is also observed in the Commission Internationale de l'Eclairage coordinate, as shown in Supplementary Table S2 and Supplementary Fig. S6. Therefore, the color-reversal effect can be practically manipulated by a UV/VUV switch with a wavelength of ~190 nm. The PLE spectra can be classified into two types. The first type monitors the emission from the 5D4 state under 542/478 nm, and the other type monitors the emission from the 5D3 state under 435/412 nm. Two findings supporting the proposed mechanism can be discovered in the PLE spectra. First, the onset wavelength of the broad band (<200 nm) and the onset energy of Figure 3b match well with the band gap value. The coincidence of both experimental and hypothetical analysis strongly supports the mechanism. Therefore, this onset can represent a switch function in the proposed route. Moreover, this wide band within the VUV region is observed only in the 5D3-type PLE spectra, but disappears in the 5D4-type, suggesting that cross-interaction might not occur between CTS and the 5D4 state. All the excited electrons from the host-lattice VB simply emit blue light and then respond to an exponential climb on the ΣI(5D3)/I(5D4) curve. Second, the other evidence for the mechanism is the shape change of the 4f→5d transition band in the 200–280 nm range. The direct feeding to the 5D4 state is considerable and leads to a low quantity of electrons that can be accommodated in the 5D3 state. Numerous studies have reported that the excited electrons in a low-temperature environment have higher probability to populate at lower energy vibrational states of the five 5d-orbitals, leading to narrow peaks, and respond to a higher resolution for excitation detection because of the relaxation process of the thermally stable phonons at individual 5d states without cross-interacting with each other43, 44. A similar phenomenon can be observed in the 5D3-type excitation spectra. The low population of 5D3 state electrons narrows down the bandwidth of the 4f–5d excitation transition in the PLE spectrum. The reason for this is that the electron population among each 5d-orbital might be limited to the localized vibrational state distribution because of the few electrons in the 5D3 state, whereas the excited energy scans until the UV region (>200 nm). As a result, the 5d level can be clearly resolved at 211, 234 and 254 nm. Although, the Tb ions are situated in nearly octahedral coordination spheres, the crystal-field strength splits the five 5d-orbitals into three different degeneracies rather than two theoretical octahedral 5d states (e.g., eg and t2g). The detailed discussion is depicted in the supporting information. However, the excitation signal of 5D3-type rapidly increases compared with that of 5D4-type, whereas the excited energy scans until the VUV region (<200 nm). These findings demonstrate that the CTS simply interacts with the 5D3 state rather than the 5D4 state.
Conclusions
In summary, a color-reversal effect, which is a specific transition route that acts as a switch of green/blue emissions, was observed in a Ba2.89Si6O12N2:Tb0.11 phosphor. This effect is expected to be generally exhibited by all phosphors doped with Tb activators. In addition, the same effect may also be applicable to other lanthanide f–f inner-shell transition systems that are sensitive to cross-relaxation mechanisms.
References
Okazaki C, Shiiki M, Suzuki T, Suzuki K . Luminance saturation properties of PDP phosphors. J Lumin 2000; 87-89: 1280–1282.
Wu JH, Yan B . Photoluminescence intensity of YxGd1−xVO4: Eu3+ dependence on hydrothermal synthesis time and variable ratio of Y/Gd. J Alloys Compd 2008; 455: 485–488.
Schubert EF, Kim JK . Solid-state light sources getting smart. Science 2005; 308: 1274–1278.
Hashimoto T, Wu F, Speck JS, Nakamura S . A GaN bulk crystal with improved structural quality grown by the ammonothermal method. Nat Mater 2007; 6: 568–571.
Xie RJ, Hirosaki N, Sakuma K, Yamamoto Y, Mitomo M . Eu2+-doped Ca-α-SiAlON: a yellow phosphor for white light-emitting diodes. Appl Phys Lett 2004; 84: 5404–5406.
Chen YB, Gong ML, Wang G, Su Q . High efficient and low color-temperature white light-emitting diodes with Tb3Al5O12: Ce3+ phosphor. Appl Phys Lett 2007; 91: 071117.
Sakuta H, Fukui T, Miyachi T, Kamon K, Hayashi H et al. Near-ultraviolet LED of the external quantum efficiency over 45% and its application to high-color rendering phosphor conversion white LEDs. J Light Vis Environ 2008; 32: 39–42.
Fukui T, Kamon K, Takeshita J, Hayashi H, Miyachi T et al. Superior illuminant characteristics of color rendering and luminous efficacy in multilayered phosphor conversion white light sources excited by near-ultraviolet light-emitting diodes. Jpn J Appl Phys 2009; 48: 112101.
Jack KH . Sialons and related nitrogen ceramics. J Mater Sci 1976; 11: 1135–1158.
Wang CM, Pan XQ, Rühle M, Riley FL, Mitomo M . Silicon nitride crystal structure and observations of lattice defects. J Mater Sci 1996; 31: 5281–5298.
Riley FL . Silicon nitride and related materials. J Am Ceram Soc 2000; 83: 245–265.
Schnick W, Huppertz H . Nitridosilicates-a significant extension of silicate chemistry. Chem Eur J 1997; 3: 679–683.
Li WY, Xie RJ, Zhou TL, Liu LH, Zhu YJ . Synthesis of the phase pure Ba3Si6O12N2: Eu2+ green phosphor and its application in high color rendition white LEDs. Dalton Trans 2014; 43: 6132–6138.
Mikami M, Shimooka S, Uheda K, Imura H, Kijima N . New green phosphors Ba3Si6O12N2: Eu for white LED: crystal structure and optical properties. Key Eng Mater 2009; 403: 11–14.
Braun C, Seibald M, Börger SL, Oeckler O, Boyko TD et al. Material properties and structural characterization of M3Si6O12N2: Eu2+ (M=Ba, Sr)—A comprehensive study on a promising green phosphor for pc-LEDs. Chem Eur J 2010; 16: 9646–9657.
Tang JY, Chen JH, Hao LY, Xu X, Xie WJ et al. Green Eu2+-doped Ba3Si6O12N2 phosphor for white light-emitting diodes: Synthesis, characterization and theoretical simulation. J Lumin 2011; 131: 1101–1106.
Höppe HA . Recent developments in the field of inorganic phosphors. Angew Chem Int Ed 2009; 48: 3572–3582.
Setlur AA . Phosphors for LED-based solid-state lighting. Electrochem Soc Interface 2009; 18: 32–36.
Xie RJ, Hirosaki N . Silicon-based oxynitride and nitride phosphors for white LEDs—a review. Sci Technol Adv Mater 2007; 8: 588–600.
Zeuner M, Pagano S, Schnick W . Nitridosilicates and oxonitridosilicates: from ceramic materials to structural and functional diversity. Angew Chem Int Ed 2011; 50: 7754–7775.
Feldmann C, Jüstel T, Ronda CR, Schmidt PJ . Inorganic luminescent materials: 100 years of research and application. Adv Funct Mater 2003; 13: 511–516.
Blasse G, Grabmaier BC . Luminescent Materials. Springer-Verlag: Berlin. 1994.
Kim CH, Kwon IE, Park CH, Hwang YJ, Bae HS et al. Phosphors for plasma display panels. J Alloys Compd 2000; 311: 33–39.
Jüstel T, Krupa JC, Wiechert DU . VUV spectroscopy of luminescent materials for plasma display panels and Xe discharge lamps. J Lumin 2001; 93: 179–189.
Zhang SX . Vacuum-ultraviolet/visible conversion phosphors for plasma display panels. IEEE Trans Plasma Sci 2006; 34: 294–304.
Robbins DJ, Cockayne B, Lent B, Glasper JL . The mechanism of 5D3–5D4 cross-relaxation in Y3Al5O12: Tb3+. Solid State Commun 1976; 20: 673–676.
Larson AC, Von Dreele RB . Generalized Structure Analysis System (GSAS). Los Alamos National Laboratory: Los Alamos, NM. 1994 Los Alamos National Laboratory Report LAUR 86-748.
Hsu CH, Lu CH . Microwave-hydrothermally synthesized (Sr1−x−yCexTbySi2O2−δN2+μ phosphors: efficient energy transfer, structural refinement and photoluminescence properties. J Mater Chem 2011; 21: 2932–2939.
Fyfe CA, Brouwer DH . Effect of molecular oxygen on the variable-temperature 29Si MAS NMR spectra of zeolite-sorbate complexes. J Am Chem Soc 2004; 126: 1306–1307.
Li YQ, Hirosaki N, Xie RJ, Takeda T, Mitomo M . Yellow-orange-emitting CaAlSiN3: Ce3+ phosphor: structure, photoluminescence, and application in white LEDs. Chem Mater 2008; 20: 6704–6714.
Park WB, Singh SP, Pyo M, Sohn KS . Y6+x/3Si11−yAlyN20+x−yO1−x+y: Re3+ (Re=Ce3+, Tb3+, Sm3+ phosphors identified by solid-state combinatorial chemistry. J Mater Chem 2011; 21: 5780–5785.
Park K, Heo MH . Enhanced photoluminescence of GdPO4: Tb3+ under VUV excitation by controlling ZnO content and annealing temperature. J Alloys Compd 2011; 509: 9111–9115.
Wen Y, Wang YH, Liu BT, Zhang F . Luminescence properties of Ca4Y6(SiO4 6O: RE3+ (RE=Eu, Tb, Dy, Sm and Tm) under vacuum ultraviolet excitation. Opt Mater 2012; 34: 889–892.
Hölsä J, Leskelä M . Luminescence properties of terbium-activated rare-earth oxyhalides. Pys Stat Sol 1981; 103: 797–801.
Dorenbos P . Predictability of 5d level positions of the triply ionized lanthanides in halogenides and chalcogenides. J Lumin 2000; 87-89: 970–972.
Dorenbos P . The 4fn↔4fn−15d transitions of the trivalent lanthanides in halogenides and chalcogenides. J Lumin 2000; 91: 91–106.
Dorenbos P . The 5d level positions of the trivalent lanthanides in inorganic compounds. J Lumin 2000; 91: 155–176.
Zhang ZJ, Ten Kate OM, Delsing A, Van Der Kolk E, Notten PHL et al. Photoluminescence properties and energy level locations of RE3+ (RE=Pr, Sm, Tb, Tb/Ce) in CaAlSiN3 phosphors. J Mater Chem 2012; 22: 9813–9820.
Yang HC, Li CY, He H, Tao Y, Xu JH et al. VUV-UV excited luminescent properties of LnCa4O(BO3 3: RE3+ (Ln=Y, La, Gd; Re=Eu, Tb, Dy, Ce). J Lumin 2006; 118: 61–69.
Jørgensen CK . Electron transfer spectra of lanthanide complexes. Mol Phys 1962; 5: 271–277.
Su Q . Proceedings of the 2nd International Conference on Rare Earth Development and Application. Beijing: International Academic Publishers. 1991, 765–769.
Nakazawa E Fundamentals of luminescence In: Shionoya S, Yen WM editors. Phosphor Handbook. Boca Raton, FL: CRC Press. 2006.
Uheda K, Hirosaki N, Yamamoto Y, Naito A, Nakajima T et al. Luminescence properties of a red phosphor, CaAlSiN3: Eu2+, for white light-emitting diodes sensors and displays: principles, materials, and processing. Electrochem Solid State Lett 2006; 9: H22–H25.
Van Pieterson L, Reid MF, Burdick GW, Meijerink A . 4fn→4fn−15d transitions of the heavy lanthanides: experiment and theory. Phys Rev B 2002; 65: 045114.
Acknowledgements
The authors thank the Science and Technology of Taiwan (Contract No. MOST 104-2113-M-002-012-MY3, MOST 104-2119-M-002-027-MY3, MOST 104-2923-M-002-007-MY3 and MOST 104-2917-I-564-060).
Author information
Authors and Affiliations
Corresponding author
Additional information
Note: Supplementary Information for this article can be found on the Light: Science & Applications' website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lin, C., Chen, WT., Chu, CI. et al. UV/VUV switch-driven color-reversal effect for Tb-activated phosphors. Light Sci Appl 5, e16066 (2016). https://doi.org/10.1038/lsa.2016.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/lsa.2016.66
Keywords
This article is cited by
-
Synthesis and optical properties of doped CsPbCl3 nanocrystals
Journal of Nanoparticle Research (2021)
-
Energy transfer and luminescence properties of a green-to-red color tunable phosphor Sr8MgY(PO4)7:Tb3+,Eu3+
Journal of Materials Science: Materials in Electronics (2019)
-
Luminescence and thermal-quenching properties of silicate-based red-emitting K4CaSi3O9:Eu3+ phosphor
Journal of Materials Science: Materials in Electronics (2018)
