
Abstract
Tyrosine kinase inhibitor (TKI) therapies induce clinical remission with remarkable effects on chronic myeloid leukemia (CML). However, very few TKIs completely eradicate the leukemic clone and persistence of leukemic stem cells (LSCs) remains challenging, warranting new, distinct targets for improved treatments. We demonstrated that the scaffold protein AHI-1 is highly deregulated in LSCs and interacts with multiple proteins, including Dynamin-2 (DNM2), to mediate TKI-resistance of LSCs. We have now demonstrated that the SH3 domain of AHI-1 and the proline rich domain of DNM2 are mainly responsible for this interaction. DNM2 expression was significantly increased in CML stem/progenitor cells; knockdown of DNM2 greatly impaired their survival and sensitized them to TKI treatments. Importantly, a new AHI-1–BCR-ABL–DNM2 protein complex was uncovered, which regulates leukemic properties of these cells through a unique mechanism of cellular endocytosis and ROS-mediated autophagy. Thus, targeting this complex may facilitate eradication of LSCs for curative therapies.
Similar content being viewed by others


Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder that originates in hematopoietic stem cells and evolves through three stages: chronic phase (CP), accelerated phase (AP) and blast crisis (BC).1, 2, 3, 4, 5 CML and a subset of acute lymphoblastic leukemia (ALL) are caused by a BCR-ABL fusion gene with constitutively elevated tyrosine kinase (TK) activity that drives CML/ALL pathogenesis.1, 2, 3, 4, 5 ABL-specific tyrosine kinase inhibitor (TKI) monotherapies have been applied successfully in CP patients.6, 7, 8 However, most patients harbor residual leukemic cells, and disease usually recurs if TKI Imatinib (IM) treatment is discontinued.9, 10, 11 One of the major challenges is the persistence of leukemic stem cells (LSCs) with multiple unique properties that are not well understood.12, 13, 14, 15, 16, 17 Therefore, it is imperative to seek other therapeutic targets in LSCs for curative therapies.
One candidate is Ahi-1 (Abelson helper integration site-1), which was identified as a cooperative oncogene in a v-abl-induced murine model.18 Human AHI-1 has an N-terminal coiled-coil domain, a WD40-repeat domain and a SH3 domain, all mediators of protein–protein interactions.18 Interestingly, AHI-1 expression is significantly elevated in CML LSCs and the AHI-1-mediated protein complex containing BCR-ABL and JAK2 contributes to the BCR-ABL transforming ability and TKI resistance of primary CML stem/progenitor cells.19, 20, 21 We have further demonstrated that the AHI-1 SH3 domain plays a critical role in mediating TKI response/resistance in BCR-ABL+ cells and identified Dynamin-2 (DNM2) as a new AHI-1 interacting protein.22
DNM2, a large GTPase, is involved in multiple cellular activities such as endocytosis, actin cytoskeleton formation and microtubule reorganization,23, 24, 25, 26 and its deregulation has been implicated in the oncogenesis of numerous malignancies.27, 28, 29, 30, 31, 32 However, the biological relevance of DNM2 in CML pathogenesis and drug resistance is unknown.
Here we demonstrate that the interaction between AHI-1 and DNM2 is mainly ascribed to SH3-PRD recognition. DNM2 expression was significantly increased in leukemic stem/progenitor cells, and DNM2 suppression reduced survival and enhanced TKI sensitivity of BCR-ABL+ blast cells and TKI-insensitive stem/progenitor cells. Importantly, a new AHI-1-mediated protein complex containing BCR-ABL and DNM2 was identified, which is strongly implicated in the deregulation of endocytosis, ROS production and autophagy in leukemic stem/progenitor cells.
Materials and methods
Patients
Heparin-anticoagulated peripheral blood (PB) or bone marrow (BM) cells from 28 CP CML patients, none previously treated with TKIs, were studied (Supplementary Table 1). Subsequent IM responders and IM nonresponders were classified based on the European Leukemia Net guidelines (Supplementary Table 1).6, 33
Human cells
PB or BM cells were obtained from newly diagnosed patients and healthy adult donors (ALLCELLS). Informed consent was obtained in accordance with the Declaration of Helsinki, and the procedures used were approved by the Research Ethics Board at the University of British Columbia. Mononuclear cells were isolated using Lymphoprep (STEMCELL Technologies, Vancouver, BC, Canada) and CD34+ cells (>85%) were enriched immunomagnetically using the EasySep CD34 positive selection kit (STEMCELL Technologies). Purity was verified by restaining isolated cells with an allophycocyanin-labeled (APC) anti-CD34 antibody (Thermo Fisher Scientific, Waltham, MA, USA) and fluorescence-activated cell sorter analysis.
Cell cultures
BCR-ABL+ human cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Life Technologies, Carlsbad, CA, USA), 0.1 mg/ml streptomycin (Thermo Fisher Scientific), 100 U/l penicillin (Thermo Fisher Scientific) and 10−4 M β-mercaptoethanol (STEMCELL Technologies). Parental BaF3 cells, human 293T cells and primary CD34+ cells were cultured as described previously.19
DNM2 constructs and lentiviral vectors
Full-length human DNM2 and DNM2 PRDΔ were cloned into the KA391 vector through AscI and PacI restriction sites. The pGFP-C-lenti vector (OriGene), containing the non-targeting sequence or DNM2 shRNA constructs, and the pRRL-PPT-SF-GFP-pre vector were used as templates to amplify the U6 promoter-shRNAs and the SFFV promoter, respectively. The two PCR products were then mixed and used as template for the third overlapping PCR. The final PCR product was then cloned into the pRRL-PPT-SF-GFP-pre vector using NdeI (NEB) and AgeI (NEB) restriction sites. Additional details are available in Supplementary Methods.
Reagents
Imatinib, ponatinib and ABL001 were obtained from Selleckchem (Houston, TX, USA). MitMAB was obtained from Abcam (Cambridge, UK).
Western blotting and co-immunoprecipitation
Detailed procedures were described previously.18, 19, 21 Briefly, for western blotting, protein samples were separated in 8–15% SDS-PAGE, followed by transfer to a polyvinylidene difluoride membrane (EMD Millipore, Sigma-Aldrich Canada Co., Oakville, ON, Canada). The membrane was subsequently incubated with specific antibodies. For co-immunoprecipitation (co-IP), cell lysates were incubated overnight with a specific primary antibody at 4 °C, and the immune complexes were incubated with protein A or protein G bead flurry (Santa Cruz Biotechnology, Dallas, TX, USA) for 2 h at 4 °C on the second day. Beads were then resuspended in SDS-loading buffer and used for western blotting. Antibodies used are listed in Supplementary Table 2.
293T cell transfection and immunostaining
For transfection, a mixture of DNA plasmids and polyethylenimine (PEI, Polysciences Inc., Warrington, PA, USA) was added to 293T cells, followed by incubation at 37 °C for 48 h. For immunostaining, cells that grew on cover slips were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-100 (Sigma-Aldrich Canada Co.) at room temperature. Cells were then blocked with 3% BSA, followed by incubation with primary and then secondary antibodies. Finally, the slides were mounted in Prolong Gold antifade reagent (Life Technologies) with DAPI. Additional details are available in Supplementary Methods.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using TRIzol (Life Technologies) according to manufacturer’s instructions.34 During RNA precipitation, glycogen (Life Technologies) was used to visualize the RNA pellet. The RNA pellet was then dissolved in RNAse-free water (Life Technologies). Quantitative real-time PCR was performed as previously described.13 Additional details are available in the Supplementary Methods.
Lentivirus production and infection
Lentiviral production of DNM2 shRNAs in 293T cells using pGFP-C-shLenti and pRRL-PPT-SF-GFP-pre viral vectors and transduction of DNM2 shRNAs into K562, BV173 and CD34+ CML cells are described in Supplementary Methods.
Viability and apoptosis assays
Total viable cells were assessed on a Neubauer hemacytometer (Hausser Scientific, Horsham, PA, USA) using the trypan blue (Life Technologies) exclusion method. Apoptosis analysis was performed using an Apoptosis Detection kit (Thermo Fisher Scientific). After being treated with inhibitors for 24 or 48 h, the cells were pelleted and resuspended in binding buffer with PI and APC-conjugated Annexin V at room temperature for 15 min. Cells were analyzed using a FACS Calibur (BD Bioscience, San Jose, CA, USA). Total apoptotic cell numbers were calculated as the sum of ‘early’ apoptotic cells (Annexin V+ only) and ‘late’ apoptotic cells (Annexin V+/PI+).
Colony-forming cell and long-term culture-initiating cell assays
Detailed procedures were previously described.19, 21, 35 Briefly, for colony-forming cell assays, 3000 primary CD34+ CML cells were mixed with 3 ml MethoCult H4230 (STEMCELL Technologies) and growth factor cocktail in the presence or absence of inhibitors. The colonies produced were counted after 14 days incubation. For the long-term culture-initiating cell assay, CD34+ CML cells were plated onto humanized feeders in Myelocult H5100 medium (STEMCELL Technologies) containing 10−6 M solu cortef (STEMCELL Technologies) for 6 weeks. Half-medium change was performed weekly. Inhibitors were added on the first day when the assays were set up. After 6 weeks, 1 × 104 viable cells were plated for colony-forming cell assays.
Transferrin uptake assay
Cells were kept on ice for 10 min to stop endocytosis, washed twice with a HEPES-based buffer at pH 7.4 containing 20 mM glucose (Bdh Inc., Conestoga, PA, USA) and 1% BSA (referred to as wash buffer thereafter). Subsequently, cells were incubated with the wash buffer containing transferrin conjugates (Life Technologies) for 30 min at 37 oC, and then kept on ice for 10 min to terminate the reaction. Cells were plated onto poly-L-lysine-coated slides, followed by fixation with 4% paraformaldehyde at room temperature. The slides were finally mounted in Prolong Gold antifade reagent with DAPI. Slides were examined using a Nikon C1 confocal microscope (Nikon Canada Inc., Mississauga, ON, Canada) and results obtained were quantified by ImageJ.
ROS staining
Cells were incubated with CellROX Reagent (Life Technologies) in complete RPMI for 30 min at 37 oC, and plated onto poly-L-lysine-coated slides for 20 min. Fixation, mounting and image acquisition were performed as described above for the transferrin uptake assay.
Statistical analysis
Results are shown as the mean±s.e.m. of values obtained in two to three independent experiments. Differences between groups were assessed using the two-tailed Student’s t-test for paired or unpaired samples, or by ANOVA. A P-value <0.05 was considered significant.
Results
Ahi-1/AHI-1 interacts with DNM2 through SH3-PRD recognition within endosomal compartments
To dissect the interaction between Ahi-1/AHI-1 and DNM2, four Ahi-1 and DNM2 constructs were generated, including HA-tagged full-length or SH3 domain-deleted Ahi-1 and Myc-tagged full-length or proline rich domain (PRD)-deleted DNM2 (Figure 1a, left). Proximity ligation assays (PLA)36 read out by confocal microscopy analysis, which detect protein interactions at a single-molecule level, revealed strong fluorescent signals only in 293T cells co-transfected with full-length Ahi-1 and DNM2, but not in cells co-expressing Ahi-1 and DNM2 mutants, suggesting that the SH3-PRD recognition is essential for the interaction between Ahi-1/AHI-1 and DNM2 (Supplementary Figure 1). Confocal images further showed that co-localization signals were observed in the cytoplasm of cells with full-length Ahi-1 and DNM2, but barely detectable in Ahi-1/DNM2 PRDΔ co-transfected cells (Figure 1a, right). Ahi-1 SH3Δ localized to the nuclei, while full-length DNM2 or DNM2 PRDΔ was cytoplasmic, so no co-localization signals were detected (Figure 1a, right). Thus, the binding between the AHI-1 SH3 domain and DNM2 PRD is critical for the interaction between Ahi-1/AHI-1 and DNM2.

The interaction between Ahi-1 and DNM2 depends on SH3-PRD recognition within endosomal compartments. (a) Schematic representations of four Ahi-1 and DNM2 constructs, including HA-tagged full-length Ahi-1 (HA-Ahi-1), HA-tagged SH3 domain-deleted Ahi-1 (HA-SH3Δ), Myc-tagged full-length DNM2 (Myc-DNM2) and Myc-tagged proline rich domain-deleted DNM2 (Myc-PRDΔ). 293T cells co-transfected with indicated constructs were stained with anti-HA (green) and anti-Myc (red) antibodies. DAPI was used to stain the nuclei. Representative images are shown. (b, c) 293T cells co-transfected with indicated constructs were stained with anti-HA (green) and anti-EEA1 (red, b) or anti-LAMP-1 (red, c) antibodies. Images were acquired using a magnification of × 60 by confocal microscopy. The white scale bar represents 5 μm.
Observing the specific co-localization signals of Ahi-1 and DNM2 in a ‘punctate’ pattern (Figure 1a), and given that DNM2 is a major component in the endocytic pathway,37, 38, 39, 40 we investigated whether the interaction occurred in endosomal compartments. 293T cells co-expressing Ahi-1 and DNM2 mutants were co-immunostained with anti-HA/anti-EEA1 (early endosomal marker) antibodies or anti-HA/anti-LAMP1 (late endosomal marker) antibodies,41 followed by co-localization analysis. In the full-length Ahi-1/DNM2 cells, Ahi-1 co-localized with EEA1 or LAMP1 in the cytoplasm; in contrast, the co-localization signals were significantly disrupted in Ahi-1/DNM2 PRDΔ mutant cells (Figures 1b and c). Co-localization signals were not detectable in cells containing Ahi-1 SH3Δ mutant since it resided in the nucleus (Figures 1b and c). These results indicate that Ahi-1’s interaction with DNM2 occurs in endosomes.
Transcript levels of DNM2 are significantly increased in CD34+ CML stem/progenitor cells
To determine whether DNM2 is involved in the pathogenesis of CML, transcript levels of DNM2 were examined in CD34+ normal and CML cells. Interestingly, DNM2 transcripts were significantly increased in treatment-naive CD34+ cells from subsequent IM responders (n=11) and IM nonresponders (n=14) compared to CD34+ normal BM cells (n=7, P=0.013 and 0.037, Figure 2a). In addition, DNM2 expression was significantly higher in CML stem-enriched (lin−CD34+CD38−, n=8) and progenitor cells (lin−CD34+CD38+, n=8) than mature cells (lin+CD34−, n=7) from IM nonresponders (P=0.0012 and 0.0229, Figure 2a). Similar results were observed from IM responders (n=5), however the stem-enriched fraction from IM nonresponders showed a modest increase in DNM2 transcript levels compared to the same population from IM responders (Figure 2a). These results demonstrated relevance of DNM2 deregulation in primitive CML cells.

Increased expression of DNM2 in CD34+ CML stem/progenitor cells and lentiviral-mediated knockdown of DNM2 in BCR–ABL+ cells affects the JAK2/STAT5 pathway. (a) Quantitative real-time PCR analysis of the transcript levels of DNM2 in CD34+ cells purified from normal bone marrow (NBM), IM responders (IM R) and IM nonresponders (IM NR). DNM2 transcript levels were normalized to the control gene β2M, and bars represent the mean of data for each group. Comparison of the transcript levels of DNM2 in three subpopulations from IM NR (n=8, red) and IM R (n=5, blue). (b) Western blot analysis of phosphorylation and protein expression levels of DNM2 and other proteins in DNM2 knockdown K562 cells (shDNM2A and shDNM2B) and BV173 cells (shDNM2A). The densitometry values of protein expression changes are indicated as compared to SHC control.
Lentiviral-mediated knockdown of DNM2 expression strongly disturbs survival, apoptosis, and the JAK2/STAT5 pathway in BCR-ABL+ blast cells
To determine the biological significance of DNM2 in CML, lentiviral-mediated DNM2 knockdown was performed in K562 and BV173 blast cells; DNM2 expression was reduced about 90 and 45% in K562 cells with two constructs (shDNM2A and shDNM2B) and 65% in BV173 cells with shDNM2A (Figure 2b). Transduced K562 cells showed a significant decrease in viability (shDNM2A vs shDNM2B, 32% vs 79%) and increased apoptosis compared to cells transduced with a vector control (SHC), which correlated with the level of DNM2 protein suppression (Supplementary Figures 2a and b). These effects were enhanced upon IM treatment. Similarly, DNM2-knockdown BV173 cells showed a significant growth disadvantage, increased apoptosis and sensitization to TKIs (IM or dasatinib (DA)) compared to control cells (Supplementary Figures 2c and d). These observations were supported by reduced viability (~50%) and increased apoptosis in K562 and BV173 SHC cells treated with MitMAB,42 a selective DNM2 inhibitor. MitMAB treatment in DNM2-knockdown cells resulted in even greater biological consequences (Supplementary Figure 2). Knockdown of DNM2 also led to reduced phosphorylation of BCR-ABL, JAK2 and STAT5, which corresponded to DNM2 suppression levels, but unaltered p-AKT and p-ERK, as compared to SHC control cells (Figure 2b). Thus, genetic and pharmacological suppression of DNM2 significantly impaired survival of BCR-ABL+ blast cells and sensitized them to TKI treatments, possibly through the DNM2/BCR-ABL-mediated JAK2/STAT5 pathway.
Lentiviral-mediated knockdown of DNM2 expression significantly impairs survival and increases TKI sensitivity of CML stem/progenitor cells
To investigate the effects of DNM2 in primary CML stem/progenitor cells, DNM2 expression was suppressed by 80–90% using lentiviral-mediated knockdown in CD34+ cells from three IM nonresponders (Supplementary Figure 3 and Supplementary Table 1). Compared to SHC controls, DNM2-suppressed CD34+ CML cells showed significantly reduced proliferative potential with or without IM (P<0.05, Figure 3a, left). DNM2 knockdown decreased the viability of CD34+ CML cells (54%) compared to SHC control cells, and the effects were further enhanced with IM (32% vs 70%) or DA (38% vs 74%, Figure 3b, left). CD34+ CML cells with DNM2 depletion also exhibited increased apoptosis, and these effects were further enhanced with IM or DA (Figure 3b, right). DNM2 suppression reduced the colony-forming ability of CD34+ cells by 38% relative to SHC control cells, and the effects were more prominent with IM or DA treatments (Figure 4a). Importantly, long-term culture-initiating cell assays demonstrated that DNM2 depletion also impaired the long-term colony growth of LSCs (37%) relative to control cells, and further sensitized them to IM (28% vs 49%) or DA (32% vs 55%, Figure 4b). MitMAB treatments in CD34+ CML cells mimicked the biological consequences of DNM2 knockdown (Figure 3a, right). Overall, DNM2 depletion had a detrimental effect on survival of TKI-insensitive stem and progenitor cells.

Lentiviral-mediated knockdown of DNM2 impairs the survival of CD34+ CML stem/progenitor cells and sensitizes these cells to TKIs. (a) Cell proliferation of control (SHC) or DNM2-knockdown CD34+ CML cells with or without 5 μM IM (left) and MitMAB (right). (b) Cell viability (left) and apoptosis (right) assays in SHC or DNM2-knockdown CD34+ CML cells with or without 5 μM IM or 150 nM DA treatments. Values shown are the mean±s.e.m. *P<0.05, ***P<0.001.

Lentiviral-mediated knockdown of DNM2 impairs the survival of very primitive CD34+ CML stem/progenitor cells and identification of the AHI-1–BCR–ABL–DNM2 protein complex. (a) Numbers and types of colonies produced by transduction of CD34+ IM-nonresponder cells (n=3) with either a control (SHC) or shDNM2A construct in semisolid culture medium with or without 5 μM IM or 150 nM DA. (b) Long-term culture-initiating cell (LTC-IC) analysis of colony-forming cell (CFC) outputs in the same transduced cells cultured for 6 weeks in the presence of stromal cells with or without 5 μM IM or 150 nM DA. (c) Co-immunoprecipitation assays in K562 and K562 IM-resistant cells (IMR, left) and BCR–ABL-transduced and BCR–ABL/Ahi-1 co-transduced BaF3 cells (middle). Protein extracts were subjected to anti-DNM2 immunoprecipitation and then immunoblotted with anti-c-Abl antibody or anti-DNM2 antibody. 293T cells were co-transfected with HA-Ahi-1, BCR–ABL and Myc-DNM2 (or Myc-DNM2 PRDΔ) or HA-Ahi-1 SH3Δ, BCR–ABL and Myc-DNM2 (or Myc-DNM2 PRDΔ, right). Cells were stained with anti-Myc (red), anti-HA (purple) and anti-c-Abl (green) antibodies. DAPI was used to stain the nuclei. Images were acquired using a magnification of × 60 by confocal microscopy. The white scale bar represents 5 μm.
Identification of a new AHI-1–BCR-ABL–DNM2 protein complex and BCR-ABL phosphorylates DNM2 in BCR-ABL+ cells
Co-IP experiments revealed a complex between BCR-ABL and DNM2 in K562 and K562 IM-resistant cells (K562 IMR) after immunoprecipitation with an anti-DNM2 antibody (Figure 4c, left). This interaction was confirmed in BCR-ABL-transduced BaF3 cells. Strikingly, this interaction was enhanced in BCR-ABL-transduced cells co-expressing full-length Ahi-1; in contrast, the interaction was not observed in BCR-ABL/Ahi-1 SH3Δ co-transduced cells (Figure 4c, middle). This was supported by the observation of strong co-localization signals in 293T cells co-expressing full-length HA-Ahi-1, BCR-ABL and Myc-DNM2, while the co-localization between Ahi-1, BCR-ABL and DNM2 PRDΔ or between Ahi-1 SH3Δ, BCR-ABL and DNM2/DNM2 PRDΔ was barely detectable (Figure 4c, right). Co-IP experiments revealed that IM treatment greatly reduced endogenous p-DNM2 levels in K562 and K562 IMR cells compared to the same cells without IM treatment (>90%, Figure 5a, left), suggesting that BCR-ABL phosphorylated DNM2 in these cells. Furthermore, BCR-ABL-transduced human UT7 cells also exhibited significantly reduced DNM2 phosphorylation upon IM treatment, but in the same cells transduced with a BCR-ABL–T315I mutant that prevents TKI binding, IM treatment did not decrease, but rather enhanced DNM2 phosphorylation (Figure 5a, right and Supplementary Figure 4). Most interestingly, treatment of the BCR-ABL–T315I mutant cells with ponatinib, a new TKI designed to specifically target the BCR-ABL–T315I mutant, resulted in significantly reduced phosphorylation of BCR-ABL and DNM2 (Figure 5d).43 The result was further supported by treatment of these cells with ABL001, which specifically binds to the myristoyl pocket of the BCR-ABL protein and inhibits most BCR-ABL mutation forms, including the T315I mutation.44 In addition, in BCR-ABL-negative BaF3 cells, tyrosine phosphorylation of DNM2 was not altered, with or without IM (Figure 5b). In 293T cells co-expressing BCR-ABL and Myc-DNM2, IM also reduced p-DNM2 levels (Figure 5c). These results provide strong evidence that AHI-1 bridges BCR-ABL and DNM2 together to form a protein complex, ‘AHI-1–BCR-ABL–DNM2 (ABD)’, which likely leads to aberrant activation of DNM2 through tyrosine phosphorylation in primitive CML cells.

Western blot analysis of the AHI-1–BCR–ABL–DNM2 protein complex, with DNM2 phosphorylation by BCR–ABL. (a, b) Co-immunoprecipitation assays in various BCR–ABL+ cell lines with or without IM for 24 h. Parental BaF3 cells were cultured with mIL-3 (10 ng) but not Ahi-1 or BCR–ABL-transduced BaF3 cells. All protein extracts were immunoprecipitated with anti-DNM2 antibody and immunoblotted with a pan-anti-p-Tyr antibody (4G10) or anti-DNM2 antibody. (c) Co-immunoprecipitation assays in BCR–ABL/Myc-DNM2 co-transfected 293T cells cultured with or without 5 μM IM for 24 h. Protein extracts were immunoprecipitated with anti-Myc antibody and then immunoblotted with a pan-anti-p-Tyr antibody (4G10) or anti-Myc antibody. (d) Co-immunoprecipitation assays of UT7 BCR–ABL T315I (UT7 B/A T315I) cells with or without ponatinib (20 nM) or ABL001 (4 μM) treatment for 24 h. All protein extracts were immunoprecipitated with an anti-DNM2 antibody and immunoblotted with a pan-anti-p-Tyr antibody (4G10).
The ABD complex enhances transferrin uptake in CML stem/progenitor cells
To investigate whether the ABD complex deregulates endocytosis in BCR-ABL+ blast cells and CML stem/progenitor cells, the transferrin uptake assay, which assesses the efficiency of endocytosis based on the amount of transferrin that accumulates inside cells, was performed.45 Compared to BCR-ABL-transduced BaF3 cells, cells co-expressing BCR-ABL and Ahi-1 exhibited significantly increased transferrin uptake (3.3-fold, Supplementary Figure 5a). However, the higher transferrin signals were strongly diminished in BCR-ABL/Ahi-1 SH3Δ co-transduced cells (1.8-fold). DNM2 knockdown significantly reduced transferrin accumulation in K562 and BV173 cells (~40 and 60%, Supplementary Figures 5b and 6a). Interestingly, IM-nonresponder CD34+ cells with DNM2 knockdown also exhibited impaired transferrin uptake (Figure 6a). Additive effects of IM in reducing intracellular transferrin signals were further observed in DNM2-knockdown BV173 or CD34+ CML cells (Supplementary Figure 6a and Figure 6a). Similarly, MitMAB, alone or in combination with IM, mimicked the results of DNM2 knockdown in CD34+ CML cells obtained from the same IM-nonresponder patients (Figure 6b). It was interesting to observe that CD34+ cells from IM responders also showed reduced intracellular transferrin signals after MitMAB and IM treatment (Supplementary Figure 6b). Thus, biological manipulation of each component of the ABD complex directly affects transferrin uptake, suggesting a new role for this complex in modulating endocytosis in CML stem/progenitor cells.

Suppression of DNM2 or inhibition of BCR–ABL affects transferrin uptake in CD34+ CML cells from IM nonresponders. CD34+ CML cells transduced with either control (SHC) or shDNM2A and cultured for 24 h (a) with or without IM or (b) CD34+ CML cells from the same patients treated with IM or MitMAB alone or in combination were stained with Alexa Fluor 647-conjugated transferrin and transferrin uptake was determined by confocal microscopy. Intracellular transferrin signals were quantified and normalized to SHC control cells or untreated cells (n=3 per group, respectively). The white scale bar represents 50 μm. Values shown are the mean±s.e.m. **P<0.01.
The ABD complex increases ROS production in CML stem/progenitor cells
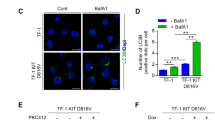
We next examined whether the ABD complex contributes to ROS overproduction in BCR-ABL+ blast cells and CD34+ CML cells. Compared to BCR-ABL-transduced BaF3 cells, co-expression of full-length Ahi-1 significantly increased ROS production (2.6-fold), while increased ROS production was not observed in Ahi-1 SH3Δ mutant cells (Supplementary Figure 7a). Knockdown of DNM2 in K562 and BV173 cells significantly reduced ROS production relative to SHC control (71 and 50% in K562 and 74% in BV173 cells, Supplementary Figures 7b and 8a). A reduction in ROS production (~60%) was also observed in IM-nonresponder CD34+ cells with DNM2 suppression (Figure 7a). Interestingly, IM treatment greatly attenuated ROS production in DNM2-knockdown BV173 and CD34+ CML cells (91% and 80%, respectively, Supplementary Figure 8a and Figure 7a). These results were further supported by treatment with MitMAB, alone or in combination with IM, in CD34+ CML cells obtained from the same IM-nonresponder patients, as well as IM responders (Figure 7b and Supplementary Figure 8b). These results suggest that the ABD protein complex may contribute to excessive generation of ROS in primitive CML cells.

Suppression of DNM2 or inhibition of BCR–ABL affects ROS production in primary CD34+ CML cells from IM nonresponders. (a, b) ROS production was determined using CellROX deep red reagents in CD34+ CML cells transduced with either a control (SHC) or shDNM2A and cultured (a) with or without IM or (b) CD34+ CML cells from the same patients treated with IM or MitMAB alone or in combination. Intracellular ROS accumulation was quantified and normalized to the signals detected in SHC or untreated control cells by confocal microscopy (n=3 per group, respectively). Representative images are shown. The white scale bar represents 50 μm. Values shown are the mean±s.e.m. *P<0.05, **P<0.01.
The ABD complex deregulates the autophagy process in CML stem/progenitor cells
Since DNM2 plays a direct role in autophagy regulation39, 46, 47 and both endocytosis and ROS production can promote autophagy activation,48, 49, 50, 51 we investigated the biological influences of the ABD protein complex on autophagy. Protein levels of four key autophagy proteins, including ULK-152 Beclin-1,53 LC3-I/LC3-II54, 55 and p62,56 were assessed in CML cells. In BCR-ABL/Ahi-1-co-transduced BaF3 cells, ULK-1, Beclin-1 and LC3-II were increased in comparison to BCR-ABL-transduced and BCR-ABL/Ahi-1 SH3Δ-cotransduced cells, while p62 was reduced (Supplementary Figure 9), providing evidence of activated autophagy. Compared to SHC controls, DNM2-knockdown K562 and BV173 cells both exhibited decreased ULK-1, Beclin-1 and LC3-II, but increased p62, suggesting decreased autophagy (Supplementary Figure 9). More importantly, knockdown of DNM2 in IM-nonresponder CD34+ cells (n=3, Supplementary Table 1) resulted in reduced ULK-1, Beclin-1 and LC3-II, but accumulated p62 (~2-fold, Figure 8a). Similarly, MitMAB treatment decreased autophagic flux in CD34+ CML stem/progenitor cells from the same patient samples, as measured with a specific Cyto-ID green detection assay, as previously reported35 (Supplementary Figure 10). Overall, these findings show a strong correlation in endocytosis and ROS production with Ahi-1 and DNM2 expression in BCR-ABL+ primitive cells.

The effects of suppression of DNM2 on key autophagy regulators in primary CD34+ CML cells and a model of biological functions of the ABD complex in CML cells. (a) Western blot analysis of ULK-1, Beclin-1, LC3-II and p62 in CD34+ IM-nonresponder cells (n=3) with knockdown of DNM2 as indicated. The densitometry values of protein expression changes are indicated. Bar graph represents the quantification of the protein levels of ULK-1, Beclin-1, LC3-II and p62 relative to Actin and SHC controls in DNM2 knockdown CD34+ CML cells. Values shown are the mean±s.e.m. **P<0.01. (b) Model of the mechanism by which the ABD protein complex deregulates three essential cellular activities—endocytosis, ROS production and autophagy—in CML stem/progenitor cells, resulting in increased LSC survival and genomic stability, but reduced TKI response of these cells. Knockdown or pharmaceutical inhibition of DNM2 activities and TKI treatments to destabilize this complex perturbs these key cellular properties.
Discussion
Understanding the molecular action of the BCR-ABL oncoprotein led to the development of small-molecule inhibitors for effective treatment of early phase CML, but disease eradication, particularly in AP and BC, will require new approaches. In this study, we uncovered a protein complex that deregulates three essential cellular activities—endocytosis, ROS production and autophagy—in BCR-ABL+ blast cells and CML stem/progenitor cells, thereby modulating the properties of LSCs and TKI resistance (Figure 8b).
Evidence indicates that deregulated DNM2 is involved in development and progression of many types of malignancies. In solid tumors, deregulation of DNM2 activity exhibits strong associations with metastatic and invasive phenotypes.27, 28, 30, 31 In T-cell leukemias, defective DNM2 leads to inactivation of T cells, so that the impaired immune surveillance fails to detect and eliminate neoplastic formations.29, 32 We have now demonstrated an oncogenic role for DNM2 in CML by showing that DNM2 is significantly upregulated in leukemic stem/progenitor cells, while DNM2 knockdown in BCR-ABL+ blast cells and CD34+ IM-nonresponder cells impairs survival and sensitizes these cells, including LSCs, as defined by long-term culture-initiating cell assays, to TKI treatments. Moreover, similar phenotypes were recapitulated in these cells upon DNM2 inhibitor treatment. These results underscore the significance of DNM2 in maintaining the functional capabilities of TKI-insensitive stem/progenitor cells and contributing to TKI resistance. Further studies should test this potential in animal models to investigate whether the concurrent inhibition of DNM2 and BCR-ABL could be a more effective approach to target the rare LSC population and prevent disease progression, once more specific and suitable DNM2 inhibitors are available for in vivo pre-clinical studies.
Mechanistically, one of the most important findings of this study was the identification of the ABD protein complex, within which BCR-ABL phosphorylates/activates DNM2. Strikingly, our study demonstrated that AHI-1 recruits DNM2 to BCR-ABL through its SH3 domain, by showing that the BCR-ABL–DNM2 interaction was enhanced in Ahi-1/BCR-ABL-co-transduced cells compared to BCR-ABL-transduced cells, and undetectable in BCR-ABL/Ahi-1 SH3Δ mutant cells (Figure 4c). Moreover, in four different cell line model systems, IM treatment resulted in decreased levels of p-DNM2, which was not observed in BCR-ABL-negative control cells or in BCR-ABL T315I mutant cells (Figure 5). This eliminated the possibility that other kinases targeted by IM were responsible for DNM2 phosphorylation in BCR-ABL+ cells. Interestingly, increased DNM2 phosphorylation was observed in BCR-ABL–T315I mutant cells, possibly because IM treatment further enhanced the kinase activity of BCR-ABL, leading to more DNM2 phosphorylation (Figure 5 and Supplementary Figure 4).57, 58 This hypothesis was further supported by the observation that DNM2 phosphorylation was strongly reduced in the BCR-ABL–T315I mutant cells treated with new TKIs, ponatinib and ABL001, designed to specifically target the BCR-ABL–T315I mutant (Figure 5d).43, 44 Interestingly, a recent study reported that DNM2 was indeed identified, via tandem affinity purifications and MS analysis, as being part of the BCR-ABL protein network.59 Thus, these findings highlight the functional importance of the ABD complex in BCR-ABL signaling and mediation of TKI response/resistance.
Strong evidence has implicated the deregulation of endocytosis in cancer development and progression.60, 61, 62 However, to our knowledge, changes in the endocytotic process have not been linked to the pathogenesis of CML and drug resistance. This study provides the first evidence that BCR-ABL+ blast cells and CML stem/progenitor cells exhibit deregulated endocytosis, mediated by the ABD protein complex. Opposite to overexpression of full-length Ahi-1, DNM2 suppression in BCR-ABL+ cells and TKI-insensitive stem/progenitor cells led to reduction in transferrin uptake (Supplementary Figures 5 and 6 and Figure 6), which is expected, given the role of DNM2 in regulation of endocytosis. Notably, IM treatment inhibited transferrin uptake, suggesting that the BCR-ABL kinase activity is also involved in endocytosis. One possible mechanism is that BCR-ABL mediates DNM2 activation, facilitated by AHI-1 (Figures 4 and 5).
It has been reported that BCR-ABL-mediated ROS overproduction causes genomic instability, leading to TKI resistance and disease progression in cell line model systems.63, 64, 65 However, specific molecular mechanisms that underpin this process are still not clear. One potential mechanism for ROS overproduction is the aberrant activation of NADPH oxidase (Nox),66, 67 whose primary function is to generate ROS. One study from Singleton et al. demonstrated that in lung endothelial cells, DNM2, activated by c-ABL phosphorylation under hypoxic conditions, promotes the activation of Nox2, leading to ROS production.68 In this study, we demonstrated that the ABD protein complex contributed to enhanced ROS production in CML stem/progenitor cells, which further explained previous observations that BCR-ABL+ cells or CML LSCs are highly unstable and generate multiple mutations both in vitro and in vivo, contributing to their insensitivity to TKIs.12, 13, 63 Thus, it is possible that highly expressed AHI-1 mediates the complex formation, ensuring efficient activation of DNM2 by BCR-ABL, thereby constantly promoting ROS generation.
Recent studies indicate that TKIs trigger the activation of autophagy, which functions as a pro-survival mechanism for CML cells to withstand TKI-induced cytotoxicity.69, 70 In this study, we hypothesized that the ABD protein complex regulates the initiation of autophagy by directly enhancing DNM2 activity or indirectly deregulating endocytosis and ROS production. Indeed, we demonstrated that Ahi-1 overexpression and DNM2 suppression exhibited opposite effects on expression of key autophagy regulators, which highly correlated with the changes observed in endocytosis and ROS production (Figures 6–8). We recently showed that the expression levels of key autophagy mediators, including Beclin-1, are significantly increased in CML stem/progenitor cells from IM-nonresponders as compared to normal bone marrow cells or IM responders.35 Furthermore, it was reported that basal autophagy was essential for BCR-ABL-mediated leukemogenesis.71 Interestingly, it has also been reported that IM-induced autophagy is associated with inhibition of the BCR-ABL/PI3K/AKT/FOXO4/ATF5/mTOR pathway.72 It would be interesting to further investigate if there is a potential link between this critical pathway and the ABD complex in regulating the autophagy process in CML. Nevertheless, this study suggests a new mechanism for how primitive CML cells maintain relatively higher basal level of autophagy compared to their normal counterparts, as a pro-survival pathway.
In conclusion, this study provided new insights into a novel mechanism causing deregulation of endocytosis, ROS production and autophagy in primitive CML cells, and suggests that dismantling the ABD protein complex by the dual inhibition of DNM2 and BCR-ABL could simultaneously impair three leukemic functions of LSCs and their progenitor cells, eventually leading to their eradication.
References
Druker BJ, O'Brien SG, Cortes J, Radich J . Chronic myelogenous leukemia. Hematol Am Soc Hematol Educ Program 2002, 111–135.
Sawyers CL . Chronic myeloid leukemia. N Engl J Med 1999; 340: 1330–1340.
Savona M, Talpaz M . Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008; 8: 341–350.
Perrotti D, Jamieson C, Goldman J, Skorski T . Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest 2010; 120: 2254–2264.
Jiang X, Smith C, Eaves A, Eaves C . The challenges of targeting chronic myeloid leukemia stem cells. Clin Lymphoma Myeloma 2007; 7 (Suppl 2): S71–S80.
Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013; 122: 872–884.
Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–2417.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344: 1031–1037.
Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11: 1029–1035.
Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007; 109: 58–60.
Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Bartley PA et al. Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia 2010; 24: 1719–1724.
Jiang X, Zhao Y, Smith C, Gasparetto M, Turhan A, Eaves A et al. Chronic myeloid leukemia stem cells possess multiple unique features of resistance to BCR-ABL targeted therapies. Leukemia 2007; 21: 926–935.
Jiang X, Saw KM, Eaves A, Eaves C . Instability of BCR-ABL gene in primary and cultured chronic myeloid leukemia stem cells. J Natl Cancer Inst 2007; 99: 680–693.
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013; 121: 1824–1838.
Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ . Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest 2011; 121: 396–409.
Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002; 99: 319–325.
O'Hare T, Zabriskie MS, Eiring AM, Deininger MW . Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer 2012; 12: 513–526.
Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P . Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J Virol 2002; 76: 9046–9059.
Chen M, Gallipoli P, DeGeer D, Sloma I, Forrest DL, Chan M et al. Targeting primitive chronic myeloid leukemia cells by effective inhibition of a new AHI-1-BCR-ABL-JAK2 complex. J Natl Cancer Inst 2013; 105: 405–423.
Jiang X, Zhao Y, Chan WY, Vercauteren S, Pang E, Kennedy S et al. Deregulated expression in Ph+ human leukemias of AHI-1, a gene activated by insertional mutagenesis in mouse models of leukemia. Blood 2004; 103: 3897–3904.
Zhou LL, Zhao Y, Ringrose A, DeGeer D, Kennah E, Lin AE et al. AHI-1 interacts with BCR-ABL and modulates BCR-ABL transforming activity and imatinib response of CML stem/progenitor cells. J Exp Med 2008; 205: 2657–2671.
Liu X, Chen M, Lobo P, An J, Grace Cheng SW, Moradian A et al. Molecular and structural characterization of the SH3 domain of AHI-1 in regulation of cellular resistance of BCR-ABL(+) chronic myeloid leukemia cells to tyrosine kinase inhibitors. Proteomics 2012; 12: 2094–2106.
Diatloff-Zito C, Gordon AJ, Duchaud E, Merlin G . Isolation of an ubiquitously expressed cDNA encoding human dynamin II, a member of the large GTP-binding protein family. Gene 1995; 163: 301–306.
Durieux AC, Prudhon B, Guicheney P, Bitoun M . Dynamin 2 and human diseases. J Mol Med 2010; 88: 339–350.
Gu C, Yaddanapudi S, Weins A, Osborn T, Reiser J, Pollak M et al. Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J 2010; 29: 3593–3606.
Ishida N, Nakamura Y, Tanabe K, Li SA, Takei K . Dynamin 2 associates with microtubules at mitosis and regulates cell cycle progression. Cell Struct Funct 2011; 36: 145–154.
Eppinga RD, Krueger EW, Weller SG, Zhang L, Cao H, McNiven MA . Increased expression of the large GTPase dynamin 2 potentiates metastatic migration and invasion of pancreatic ductal carcinoma. Oncogene 2012; 31: 1228–1241.
Feng H, Liu KW, Guo P, Zhang P, Cheng T, McNiven MA et al. Dynamin 2 mediates PDGFRalpha-SHP-2-promoted glioblastoma growth and invasion. Oncogene 2012; 31: 2691–2702.
Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest 2008; 118: 2427–2437.
Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA . Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev Cell 2013; 24: 573–585.
Rosse C, Lodillinsky C, Fuhrmann L, Nourieh M, Monteiro P, Irondelle M et al. Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc Natl Acad Sci USA 2014; 111: E1872–E1879.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012; 481: 157–163.
Forrest DL, Jiang X, Eaves CJ, Smith CL . An approach to the management of chronic myeloid leukemia in British Columbia. Curr Oncol 2008; 15: 90–97.
Rio DC, Ares M Jr, Hannon GJ, Nilsen TW . Purification of RNA using TRIzol (TRI reagent). Cold Spring Harbor protocols 2010; 2010, pdb prot5439.
Rothe K, Lin H, Lin KB, Leung A, Wang HM, Malekesmaeili M et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014; 123: 3622–3634.
Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006; 3: 995–1000.
Gonzalez-Jamett AM, Momboisse F, Haro-Acuna V, Bevilacqua JA, Caviedes P, Cardenas AM . Dynamin-2 function and dysfunction along the secretory pathway. Front Endocrinol 2013; 4: 126.
Nicoziani P, Vilhardt F, Llorente A, Hilout L, Courtoy PJ, Sandvig K et al. Role for dynamin in late endosome dynamics and trafficking of the cation-independent mannose 6-phosphate receptor. Mol Biol Cell 2000; 11: 481–495.
Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA et al. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol 2013; 203: 315–326.
van Dam EM, Stoorvogel W . Dynamin-dependent transferrin receptor recycling by endosome-derived clathrin-coated vesicles. Mol. Biol Cell 2002; 13: 169–182.
Dunster K, Toh BH, Sentry JW . Early endosomes, late endosomes, and lysosomes display distinct partitioning strategies of inheritance with similarities to Golgi-derived membranes. Eur J Cell Biol 2002; 81: 117–124.
Quan A, McGeachie AB, Keating DJ, van Dam EM, Rusak J, Chau N et al. Myristyl trimethyl ammonium bromide and octadecyl trimethyl ammonium bromide are surface-active small molecule dynamin inhibitors that block endocytosis mediated by dynamin I or dynamin II. Mol Pharmacol 2007; 72: 1425–1439.
O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009; 16: 401–412.
Wylie A, Schoepfer J, Berellini G, Cai H, Caravatti G, Cotesta S et al. ABL001, a potent allosteric inhibitor of BCR-ABL, prevents emergence of resistant disease when administered in combination with Nilotinib in an in vivo murine model of chronic myeloid leukemia. Blood 2014; 124: 398 (abstract).
Barysch SV, Jahn R, Rizzoli SO . A fluorescence-based in vitro assay for investigating early endosome dynamics. Nat Protoc 2010; 5: 1127–1137.
Durieux AC, Vignaud A, Prudhon B, Viou MT, Beuvin M, Vassilopoulos S et al. A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet 2010; 19: 4820–4836.
Takahashi Y, Tsotakos N, Liu Y, Young MM, Serfass J, Tang Z et al. The Bif-1-Dynamin 2 membrane fission machinery regulates Atg9-containing vesicle generation at the Rab11-positive reservoirs. Oncotarget 2016; 7: 20855–20868.
Kongara S, Karantza V . The interplay between autophagy and ROS in tumorigenesis. Front Oncol 2012; 2: 171.
Lamb CA, Dooley HC, Tooze SA . Endocytosis and autophagy: shared machinery for degradation. Bioessays 2013; 35: 34–45.
Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M . Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 2015; 4: 184–192.
Tooze SA, Abada A, Elazar Z . Endocytosis and autophagy: exploitation or cooperation? Cold Spring Harb Perspect Biol 2014; 6: a018358.
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–1991.
Wirawan E, Lippens S, Vanden Berghe T, Romagnoli A, Fimia GM, Piacentini M et al. Beclin1: a role in membrane dynamics and beyond. Autophagy 2012; 8: 6–17.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720–5728.
Russell RC, Yuan HX, Guan KL . Autophagy regulation by nutrient signaling. Cell Res 2014; 24: 42–57.
Lippai M, Low P . The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int 2014; 2014: 832704.
O'Hare T, Walters DK, Stoffregen EP, Sherbenou DW, Heinrich MC, Deininger MW et al. Combined Abl inhibitor therapy for minimizing drug resistance in chronic myeloid leukemia: Src/Abl inhibitors are compatible with imatinib. Clin Cancer Res 2005; 11 (19 Pt 1): 6987–6993.
Tipping AJ, Baluch S, Barnes DJ, Veach DR, Clarkson BM, Bornmann WG et al. Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors in cells sensitive and resistant to imatinib mesylate. Leukemia 2004; 18: 1352–1356.
Brehme M, Hantschel O, Colinge J, Kaupe I, Planyavsky M, Kocher T et al. Charting the molecular network of the drug target Bcr-Abl. Proc Natl Acad Sci USA 2009; 106: 7414–7419.
Di Fiore PP . Endocytosis, signaling and cancer, much more than meets the eye. Preface. Mol Oncol 2009; 3: 273–279.
Mellman I, Yarden Y . Endocytosis and cancer. Cold Spring Harb Perspect Biol 2013; 5: a016949.
Mosesson Y, Mills GB, Yarden Y . Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 2008; 8: 835–850.
Koptyra M, Falinski R, Nowicki MO, Stoklosa T, Majsterek I, Nieborowska-Skorska M et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood 2006; 108: 319–327.
Muvarak N, Nagaria P, Rassool FV . Genomic instability in chronic myeloid leukemia: targets for therapy? Curr Hematol Malig Rep 2012; 7: 94–102.
Sattler M, Verma S, Shrikhande G, Byrne CH, Pride YB, Winkler T et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem 2000; 275: 24273–24278.
Irwin ME, Johnson BP, Manshouri R, Amin HM, Chandra J . A NOX2/Egr-1/Fyn pathway delineates new targets for TKI-resistant malignancies. Oncotarget 2015; 6: 23631–23646.
Naughton R, Quiney C, Turner SD, Cotter TG . Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009; 23: 1432–1440.
Singleton PA, Pendyala S, Gorshkova IA, Mambetsariev N, Moitra J, Garcia JG et al. Dynamin 2 and c-Abl are novel regulators of hyperoxia-mediated NADPH oxidase activation and reactive oxygen species production in caveolin-enriched microdomains of the endothelium. J Biol Chem 2009; 284: 34964–34975.
Can G, Ekiz HA, Baran Y . Imatinib induces autophagy through BECLIN-1 and ATG5 genes in chronic myeloid leukemia cells. Hematology 2011; 16: 95–99.
Ertmer A, Huber V, Gilch S, Yoshimori T, Erfle V, Duyster J et al. The anticancer drug imatinib induces cellular autophagy. Leukemia 2007; 21: 936–942.
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30: 1855–1867.
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR . BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 2011; 118: 2840–2848.
Acknowledgements
We thank the Stem Cell Assay Laboratory for processing patient samples; members of the Leukemia/BMT Program of BC and the Hematology Cell Bank of BC for providing patient samples; the Terry Fox Laboratory FACS Facility for cell sorting and STEMCELL Technologies for culture reagents. This work was supported by the Canadian Institutes of Health Research and in part by the Leukemia & Lymphoma Society of Canada and the Canadian Cancer Society (XJ). KR received a Medical Genetics Graduate Entrance Scholarship from the University of British Columbia and a BC Cancer Agency Incentive Training Award, RY is a recipient of Canada Graduate Scholarship, and TM held a postdoctoral research fellowship from the German Cancer Aid.
Author contributions
XL, KR and XJ developed the concept and designed experiments; XL performed most experiments, analyzed data and prepared figures; KR performed experiments, analyzed data for primary CML samples and prepared figures; RY, CF and MC performed some viability/apoptosis assays and co-IP experiments; TM and KH designed a lentiviral vector for transduction of primary CML cells; DLF provided clinical samples and clinical data; XL, KR and XJ wrote the manuscript and all other authors commentated on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Liu, X., Rothe, K., Yen, R. et al. A novel AHI-1–BCR-ABL–DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia 31, 2376–2387 (2017). https://doi.org/10.1038/leu.2017.108
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.108
This article is cited by
-
A novel synthetic chalcone derivative, 2,4,6-trimethoxy-4′-nitrochalcone (Ch-19), exerted anti-tumor effects through stimulating ROS accumulation and inducing apoptosis in esophageal cancer cells
Cell Stress and Chaperones (2022)
-
A review of Dynamin 2 involvement in cancers highlights a promising therapeutic target
Journal of Experimental & Clinical Cancer Research (2021)
-
Signal-transducing adapter protein-1 is required for maintenance of leukemic stem cells in CML
Oncogene (2020)
-
Expression of AHI1 Rescues Amyloidogenic Pathology in Alzheimer’s Disease Model Cells
Molecular Neurobiology (2019)
