
Abstract
This randomized, phase III, open-label, multicenter study compared carfilzomib monotherapy against low-dose corticosteroids and optional cyclophosphamide in relapsed and refractory multiple myeloma (RRMM). Relapsed and refractory multiple myeloma patients were randomized (1:1) to receive carfilzomib (10-min intravenous infusion; 20 mg/m2 on days 1 and 2 of cycle 1; 27 mg/m2 thereafter) or a control regimen of low-dose corticosteroids (84 mg of dexamethasone or equivalent corticosteroid) with optional cyclophosphamide (1400 mg) for 28-day cycles. The primary endpoint was overall survival (OS). Three-hundred and fifteen patients were randomized to carfilzomib (n=157) or control (n=158). Both groups had a median of five prior regimens. In the control group, 95% of patients received cyclophosphamide. Median OS was 10.2 (95% confidence interval (CI) 8.4–14.4) vs 10.0 months (95% CI 7.7–12.0) with carfilzomib vs control (hazard ratio=0.975; 95% CI 0.760–1.249; P=0.4172). Progression-free survival was similar between groups; overall response rate was higher with carfilzomib (19.1 vs 11.4%). The most common grade ⩾3 adverse events were anemia (25.5 vs 30.7%), thrombocytopenia (24.2 vs 22.2%) and neutropenia (7.6 vs 12.4%) with carfilzomib vs control. Median OS for single-agent carfilzomib was similar to that for an active doublet control regimen in heavily pretreated RRMM patients.
Similar content being viewed by others


Introduction
Therapeutic options are limited for patients with relapsed and refractory multiple myeloma (RRMM). Although treatment may induce remission, patients experience multiple relapses.1 Each subsequent remission becomes increasingly shorter in duration, and patients ultimately die from complications of the disease.2, 3, 4, 5 Historically, patients have survival duration of <12 months and response rates of approximately 15% at fourth relapse.3, 4, 6, 7
Treatment options for patients refractory to both the proteasome inhibitor bortezomib and an immunomodulatory agent are especially limited. In a retrospective study of 286 patients who were refractory to both bortezomib and immunomodulatory agents, median overall survival (OS) was approximately 9 months.3
Carfilzomib and pomalidomide are two treatments that are currently approved in the United States for patients with RRMM who have received prior bortezomib and either thalidomide or lenalidomide.8, 9 The approval of carfilzomib, a proteasome inhibitor that binds selectively and irreversibly to its target, was based on the overall response rate (ORR) from a phase II study in heavily pretreated patients.10 That study and other phase II studies further established the safety profile of single-agent carfilzomib.10, 11, 12, 13, 14, 15
Here we present the results of the randomized phase III study PX-171-011 (FOCUS), which investigated single-agent carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in patients with advanced RRMM. In the absence of an established standard of care, the control regimen was chosen on the basis of discussions with multiple myeloma (MM) experts and results from several small studies (20–42 patients) that showed disease response and symptom abatement using this combination as salvage therapy in advanced MM.16, 17, 18
Methods
Participants
FOCUS was a randomized, open-label, phase III study. Participants were recruited from 77 study sites in Europe, Australia, New Zealand and South Korea.
Eligible patients (age ⩾18 years) with Eastern Cooperative Oncology Group performance status 0–2 had received at least three prior treatments for MM, including bortezomib, lenalidomide or thalidomide, an alkylating agent, corticosteroids and anthracycline (for patients enrolled prior to Amendment 2), and were refractory to their most recent therapy. Patients with some impairment of bone marrow and organ function were eligible, including those with reduced platelet counts (>30 000/μl) and with creatinine clearance (CrCl) ⩾15 ml/min.
Exclusion criteria included the presence of Waldenström macroglobulinemia, immunoglobulin M myeloma, plasma cell leukemia, other malignancy in the previous 3 years, POEMS syndrome, prior carfilzomib, major surgery within 21 days prior to randomization, New York Heart Association class III/IV congestive heart failure, myocardial infarction in the previous 3 months, HIV seropositivity, active hepatitis, and significant neuropathy (grade 3 or 4, or grade 2 with pain).
The protocol was approved by each study center’s Institutional Review Board or Independent Ethics Committee. The study was conducted in accordance with the Declaration of Helsinki and was consistent with the Guidelines for Good Clinical Practice and all applicable regulatory guidelines. All patients provided written informed consent.
Procedures
Patients were randomized 1:1 using an interactive Web response system to receive either carfilzomib or control (defined as corticosteroids with optional cyclophosphamide) and were stratified by number of prior therapies (3 vs 4 vs ⩾5) and geographic region (Europe vs non-Europe). The randomization schedule was prepared using a blocked randomization scheme (block size of 4). Study investigators and patients were not blinded to treatment. The sponsor’s study team did not evaluate unblinded aggregate study results until completion of the study. Unmasked safety data were evaluated by an independent data-monitoring committee via an independent external statistical analysis group on a regular basis.
Patients were treated in 28-day cycles according to the following schedule: during cycles 1–9, patients in the carfilzomib group received carfilzomib at a starting dose of 20 mg/m2 administered intravenously over 10 min on days 1 and 2 of cycle 1, which was escalated to a target dose of 27 mg/m2 on days 8, 9, 15 and 16 of cycle 1 and continued on days 1, 2, 8, 9, 15 and 16 of cycles 2–9. During cycles 10 and beyond, carfilzomib was administered at 27 mg/m2 on days 1, 2, 15 and 16; treatment on days 8 and 9 was optional per the investigator’s discretion. During cycle 1, patients received oral (PO) and intravenous (IV) hydration (before and after dose), dexamethasone 4 mg (before dose; PO or IV), and ciprofloxacin (500 mg PO, once daily (QD)). For patients with a history of herpes zoster, valacyclovir or an equivalent antiviral medication was required.
For patients randomized to receive control, treatment consisted of a corticosteroid (prednisone 30 mg PO every other day, dexamethasone 6 mg PO every other day, or other equivalent corticosteroid (not to exceed 84 mg dexamethasone or equivalent per 28-day cycle)). Patients could receive optional cyclophosphamide 50 mg PO, QD (1400 mg maximum per cycle) per the investigator’s discretion.
Treatment was continued until confirmed progressive disease, unacceptable toxicity or consent withdrawal. Dose reductions of carfilzomib to 20, 15 or 11 mg/m2 were permitted, as necessary, to manage toxicity.
Objectives
The primary endpoint of the study was OS, which was defined as the time from randomization to death from any cause. Secondary endpoints included progression-free survival (PFS), which was defined as the time from randomization to confirmed progressive disease or death from any cause; ORR, which was defined as the proportion of patients who achieved partial response or better according to the International Myeloma Working Group criteria;19 duration of response, which was defined as the time from the start date of achieving a partial response or better until confirmed progressive disease or death from any cause; clinical benefit rate, which was defined as ORR with the addition of minimal response according to European Group for Blood and Marrow Transplant criteria;20 disease control rate, which was defined as clinical benefit rate plus stable disease lasting ⩾8 weeks; and safety.
Assessments
Patients were evaluated for disease response and progression (as determined by investigator assessment) on day 1 of each cycle. Treatment-emergent adverse events were assessed at each visit according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.0 and were coded using the Medical Dictionary for Regulatory Activities v15.1. Treatment-emergent adverse events were defined as adverse events (AEs) that started on or after the first administration of study treatment and within 30 days of the last administration of study treatment. All available laboratory data were reported according to International System of Units. Results of selected laboratory analyses were summarized descriptively and graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, v4.0. An independent data-monitoring committee reviewed the study data on an ongoing basis. A preplanned interim efficacy analysis took place after approximately 75% of the OS events had occurred, and the independent data-monitoring committee recommended that the study be continued until enough events had occurred to perform the final analysis.
Statistical analyses
The study had a planned enrollment of 302 patients; it was estimated that 253 OS events would provide 80% power to detect a 30% reduction in the risk of death (that is, hazard ratio (HR) of 0.70) with an overall one-sided alpha of 0.025 for carfilzomib compared with control, if one interim and final analysis were planned. The interim analysis was scheduled to occur after approximately 75% of the planned total OS events were reported. An O’Brien-Fleming-type alpha-spending function determined the monitoring boundaries for early stopping for efficacy so that the overall Type I error was less than or equal to 0.025 (one-sided). The significance level was adjusted per actual number of events observed using a Lan-DeMets implementation of the O’Brien-Fleming group-sequential procedure to account for multiple analyses of the primary endpoint (one interim analysis and one final analysis).
The assumptions for the performance of the control arm were estimated from available information from retrospective analyses, as there were no randomized trial results in advanced RRMM, and were further supported by review publications that estimated a median OS of approximately 6 months for patients with multiply relapsed MM.6, 7 The assumption for the carfilzomib group (median OS of approximately 8.6 months) was derived from phase II results (median OS of 15.4 months),10 while adjusting for the more advanced disease characteristics expected in the FOCUS study compared with previous phase II trials.
All efficacy analyses were based on the intent-to-treat population, which consisted of all randomized patients. A stratified log-rank test was used as primary inference to compare time to event outcomes such as OS and PFS between the two treatment groups. One-sided P-values for OS and PFS were from the stratified log-rank test with number of previous therapies (3 vs 4 vs ⩾5) and geographical region (Europe vs non-Europe) as stratification factors. The distribution of OS and PFS were summarized using the Kaplan–Meier method per arm. Hazard ratios and corresponding 95% confidence intervals (CIs) were calculated using a stratified Cox proportional hazards model. One-sided P-values for ORR, clinical benefit rate and disease control rate were from the Cochran–Mantel–Haenszel chi-squared test using the same randomization stratification factors as above. Adverse events were summarized using descriptive statistics. Relative dose intensity was calculated as the actual dose intensity divided by the planned dose intensity.
This study is registered with Clinicaltrials.gov, identifier NCT01302392.
Changes in methods after trial commencement
In order to adhere to guidance from the Committee for Medicinal Products for Human Use to design a study with survival as the primary endpoint, the primary endpoint was changed from PFS to OS. This was accompanied by an increase in sample size from 84 to 302 in order to provide 80% power to detect a 30% reduction in the risk of death for carfilzomib over control. A regional stratification variable was added to control for the additional sites and countries that were added for the increased sample size. Since OS became the primary endpoint, investigator’s assessment of response and progression were used to determine the respective secondary efficacy endpoints, instead of the Independent Review Committee.
To align with International Myeloma Working Group guidelines, patients with minimal response or stable disease lasting at least 8 weeks were considered to have achieved disease control (originally 6 weeks).
An inclusion criterion that required patients to have received prior treatment with an anthracycline was removed in order to expand study access to patients being managed per current standard of care (which does not support the routine use of anthracyclines). An exclusion criterion that excluded patients with any contraindications to required concomitant drugs or supportive treatments was added to ensure patient safety.
Results
Patients and enrollment
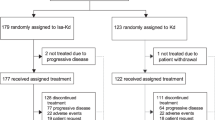
From September 2010 to October 2012, 403 patients from 77 study sites in Europe, Australia, New Zealand and South Korea were screened for eligibility. A total of 315 patients were randomized into the study and comprised the intent-to-treat population (Figure 1); 157 patients were randomized to the carfilzomib group and 158 were randomized to the control group. The safety population included the 310 patients who received at least one dose of study drug (carfilzomib, n=157; control, n=153).

Random assignment and follow-up.
The required number of events to conduct the final efficacy analysis per protocol was reached on 10 July 2014 and this is the data cutoff date used for this analysis. At data cutoff, six patients in each of the carfilzomib and control groups were alive and receiving study treatment. Median follow-up time for OS was 27.8 months (95% CI 24.6–33.7) and 29.8 months (95% CI 24.3–33.6) in the carfilzomib and control groups, respectively.
Baseline characteristics were generally well-balanced between treatment groups, with the exception being that more patients in the carfilzomib group had urine protein electrophoresis-measurable only disease (17 vs 9%) and light-chain proteinuria (18 vs 10%) only (Table 1). The overall study population had poor prognostic characteristics, including International Staging System stage III and Eastern Cooperative Oncology Group performance status ⩾2 in 39 and 20% of patients, respectively. Patients received a median of five prior regimens of therapy (range, 3 to 17) and a median of eight prior antimyeloma agents (range, 4 to 17). Sixty-three percent of patients were refractory to both bortezomib and an immunomodulatory agent (Table 2).
Efficacy
Median treatment duration was 16.3 weeks (range, 0.3 to 138.4) in the carfilzomib group and 10.7 weeks (range, 0.4 to 138.3) in the control group. Ninety-five percent of patients in the control group received cyclophosphamide in addition to corticosteroids. Relative carfilzomib and corticosteroid dose intensity was 99.9% in both the carfilzomib group and the control group.
The data met the assumptions of the statistical tests used. The similarity of variances between treatment groups for time-to-event outcomes was not compared as the variability of the per-arm estimate (for example, median) depends on the number of events, which is determined by efficacy.
The HR for OS in the carfilzomib group compared with the control group was 0.975 (95% CI 0.760–1.249; one-sided P=0.4172). As the study did not meet the primary objective of carfilzomib superiority over control, based on the hierarchical testing procedure to control multiplicity, all the P-values for secondary endpoints were multiplicity unadjusted. Median OS was 10.2 months (95% CI 8.4–14.4) with carfilzomib compared with 10.0 months (95% CI 7.7–12.0) with control (Figure 2). OS rates at 12 and 18 months were 48 and 34% in the carfilzomib group vs 42 and 30% in the control group, respectively (Table 3).

Kaplan−Meier estimates of (a) OS and (b) PFS.
Median PFS was 3.7 months (95% CI 2.8–4.2) in the carfilzomib group compared with 3.3 months (95% CI 2.2–5.2) in the control group (HR 1.091; 95% CI 0.843–1.410; one-sided P=0.2479) (Figure 2). PFS rates at 12 and 18 months were 14 and 7% with carfilzomib vs 17 and 12% with control, respectively (Table 3).
In the carfilzomib group, 66.9% of patients received any subsequent antimyeloma therapy compared with 62.0% of patients in the control group (Supplementary Table 1). A trend was observed of patients in the control group starting next antimyeloma therapy sooner than in the carfilzomib group, which led to more patients in the control group being censored for PFS. An exploratory analysis of PFS that treated the receipt of next antimyeloma therapy as a PFS event was consistent with the primary PFS analysis (Supplementary Table 2).
The ORR was higher in the carfilzomib group compared with the control group (19.1 vs 11.4%, respectively; odds ratio, 1.84; 95% CI 0.97–3.49; one-sided P=0.0305) (Table 3). A trend was observed for higher ORRs in the carfilzomib group among patients sensitive to bortezomib, refractory to bortezomib, or refractory to both bortezomib and an immunomodulatory agent (Supplementary Table 3). For all patients achieving at least partial response, the median duration of response was 7.2 months (95% CI 4.6–12.0) in the carfilzomib and 9.5 months (95% CI 3.7—not estimable) in the control group. The clinical benefit rate was 31.2 vs 20.9% in the carfilzomib vs control groups (odds ratio, 1.70; 95% CI 1.02–2.85; one-sided P=0.0212), and the disease control rate was 75.8 vs 67.7%, respectively.
Safety
Incidence rates of any-grade treatment-emergent adverse events were similar between the carfilzomib (98% of patients) and control (94%) groups (Table 4). Serious AEs (carfilzomib group, 59%; control group, 51%) and grade 5 AEs (carfilzomib group, 19%; control group, 22%) occurred at similar rates in both groups. Deaths due to AEs occurred in 10% of patients in the carfilzomib group compared with 12% in the control group (Supplementary Table 4). In the carfilzomib group, 5% of patients had their dose reduced at least once due to an AE, 1% had their dose interrupted due to an AE, 13% had their dose delayed due to an AE. Discontinuation due to an AE occurred in 15% of patients in the carfilzomib group and 20% of patients in the control group.
Grade ⩾3 AEs were similar between groups, with the exception of acute renal failure (carfilzomib, 8%; control, 3%) and pneumonia (carfilzomib, 6%; control, 12%) (see Table 4). Common grade ⩾3 nonhematologic AEs in the carfilzomib group were renal failure/acute renal failure (13%), disease progression (11%), and pneumonia (6%); common grade ⩾3 nonhematologic AEs in the control group were pneumonia (12%) and disease progression (12%). Hypertension occurred in 15% of carfilzomib patients and 6% of control patients.
Grouped renal failure AEs (that is, azotemia, oliguria, renal failure, acute renal failure and renal impairment) occurred more frequently in patients in the carfilzomib group compared with the control group (24 vs 9%). Overall, these events occurred more frequently in patients with lower CrCl at baseline (36% of all patients with baseline CrCl <30 ml/min, 23% of all patients with baseline CrCl of 30–50 ml/min and 12% of all patients with baseline CrCl ⩾50 ml/min). Furthermore, a majority of the patients who reported grouped acute renal failure AEs had urine protein electrophoresis-measurable disease (carfilzomib group, 74%; control group, 71%) compared with 26 and 29%, respectively, of those without urine protein electrophoresis-measureable disease.
Discussion
Patients with advanced RRMM have poor outcomes and few treatment options as they are likely to have been exposed to all major classes of therapeutic agents and are often refractory to at least one agent. The FOCUS study investigated single-agent carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in heavily pretreated patients with RRMM. The study did not meet its primary endpoint; there was no significant improvement in OS for carfilzomib compared with control (HR for OS 0.975; 95% CI 0.760–1.249; P=0.4172). PFS was also similar between the two treatment groups.
The single-agent activity of carfilzomib was demonstrated by the ORR of 19.1%, which was nearly twice as high as that noted with the control group (11.4%; one-sided P=0.0305); the proportion of patients achieving a minimal response or better in the carfilzomib group was also higher than that in the control group (31.2 vs 20.8%, respectively).
Incidence rates of AEs were similar between groups, with the exception of an increase in the rate any-grade hypertension (15 vs 6%) and grade ⩾3 grouped renal failure events in the carfilzomib group (24 vs 9%). The increase in renal failure events may be possibly explained by the inclusion of late-line patients with heavy disease burden, with events occurring more frequently in patients with low baseline CrCl. Notably, no increase in peripheral neuropathy was observed with carfilzomib relative to control.
The study design was based on encouraging phase II results observed with single-agent carfilzomib, which showed an ORR of 24% and a median OS of 15.2 months in patients who had received a median of five prior treatment lines (range, 1-20).10 The efficacy results observed in the present study were comparable to those reported by Siegel et al,10 while the comparatively shorter median OS of 10.2 months was likely due to differences in enrollment criteria between the two studies. In the phase II PX-171-003-A1 study, patients must have received at least two prior regimens, had platelet counts ⩾50 000/mm3, and CrCl ⩾30 ml/min; the FOCUS study specified that patients had to have received at least three prior regimens, have platelet counts ⩾30 000/mm3, and CrCl ⩾15 ml/min.
Notably, the FOCUS study compared single-agent carfilzomib against an active doublet control regimen. The addition of dexamethasone to carfilzomib should be considered, particularly in patients with suboptimal response, a strategy that had been followed with bortezomib in the APEX study.21
Further study limitations may include the open-label nature of the study, which may have led to more patients in the control group initiating their next antimyeloma regimen prior to disease progression or death (that is, censored for PFS due to start of next therapy). It is also worth noting that treatments for RRMM are often most effective in standard-risk patients; enrolling patients with advanced disease, such as those with very poor Eastern Cooperative Oncology Group performance status, renal function and/or low platelet counts, poses a significant hurdle for any single-agent therapy to demonstrate clinical benefit.
In conclusion, the phase III FOCUS study did not meet its primary end point for OS, as the active control arm of corticosteroids and optional cyclophosphamide (in which 95% of patients received cyclophosphamide) performed better than expected and demonstrated activity in this patient population. This active control arm merits further evaluation in patients with RRMM. The safety profile of single-agent carfilzomib was generally consistent with what has been reported previously in heavily pretreated patients with MM, with the exception of increased renal events.10 Carfilzomib combined with dexamethasone and/or other agents remains an active treatment option for patients with advanced MM,22 based on positive results from phase III studies in RRMM. The phase III study ASPIRE (NCT01080391) demonstrated that the addition of carfilzomib to lenalidomide-dexamethasone led to a HR for PFS of 0.69 (95% CI, 0.57–0.83) for patients with relapsed MM,23 and the phase III study ENDEAVOR (NCT01568866) met its primary endpoint of improved PFS with carfilzomib (20/56 mg/m2; 30-min infusion)-dexamethasone vs bortezomib-dexamethasone (HR 0.53; 95% CI 0.44–0.65) in patients with relapsed MM.24 Therefore, despite the lack of a survival advantage observed in late-line RRMM patients, carfilzomib remains an important component of anti-MM treatment in a variety of settings.
References
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008; 111: 2516–2520.
Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc 2004; 79: 867–874.
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012; 26: 149–157.
Durie BG, Moreau P, Sonneveld P, Morgan GJ, Lahuerta JJ, Beksac M et al. Regional differences in the treatment approaches for relapsed multiple myeloma: an IMF study. J Clin Oncol 2012; 30: 15s (abstract 8095).
Richardson PG, Sonneveld P, Schuster M, Irwin D, Stadtmauer E, Facon T et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final time-to-event results of the APEX trial. Blood 2007; 110: 3557–3560.
Anderson KC, Kyle RA, Rajkumar SV, Stewart AK, Weber D, Richardson P et al. Clinically relevant end points and new drug approvals for myeloma. Leukemia 2008; 22: 231–239.
van de Donk NW, Lokhorst HM, Dimopoulous M, Cavo M, Morgan G, Einsele H et al. Treatment of relapsed and refractory multiple myeloma in the era of novel agents. Cancer Treat Rev 2011; 37: 266–283.
Dimopoulos MA, Richardson PG, Moreau P, Anderson KC . Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol 2015; 12: 42–54.
Offidani M, Corvatta L, Caraffa P, Leoni P, Pautasso C, Larocca A et al. Pomalidomide for the treatment of relapsed-refractory multiple myeloma: a review of biological and clinical data. Expert Rev Anticancer Ther 2014; 14: 499–510.
Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012; 120: 2817–2825.
Jagannath S, Vij R, Stewart AK, Trudel S, Jakubowiak AJ, Reiman T et al. An open-label, single-arm pilot phase II study (PX-171-003-A0) of low-dose, single-agent carfilzomib in patients with relapsed and refractory multiple myeloma. Clin Lymphoma Myeloma Leuk 2012; 12: 310–318.
Vij R, Siegel DS, Jagannath S, Stewart AK, McDonagh K, Bahlis N et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012; 158: 739–748.
Vij R, Wang M, Kaufman JL, Lonial S, Jakubowiak AJ, Stewart AK et al. An open-label, single-arm, phase 2 (PX-171-004) study of single-agent carfilzomib in bortezomib-naive patients with relapsed and/or refractory multiple myeloma. Blood 2012; 119: 5661–5670.
Badros AZ, Vij R, Martin T, Zonder JA, Kunkel L, Wang Z et al. Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia 2013; 27: 1707–1714.
Siegel D, Martin T, Nooka A, Harvey RD, Vij R, Niesvizky R et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II trials. Haematologica 2013; 98: 1753–1761.
Brandes LJ, Israels LG . Weekly low-dose cyclophosphamide and alternate-day prednisone: an effective low toxicity regimen for advanced myeloma. Eur J Haematol 1987; 39: 362–368.
de Weerdt O, van de Donk NW, Veth G, Bloem AC, Hagenbeek A, Lokhorst HM . Continuous low-dose cyclophosphamide-prednisone is effective and well tolerated in patients with advanced multiple myeloma. Neth J Med 2001; 59: 50–56.
Zhou F, Guo L, Shi H, Lin C, Hou J . Continuous administration of low-dose cyclophosphamide and prednisone as a salvage treatment for multiple myeloma. Clin Lymphoma Myeloma Leuk 2010; 10: 51–55.
Durie BG, Harousseau JL, Miguel JS, Bladé J, Barlogie B, Anderson K et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20: 1467–1473.
Bladé J, Samson D, Reese D, Apperley J, Björkstrand B, Gahrton G et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Br J Haematol 1998; 102: 1115–1123.
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352: 2487–2498.
NCCNClinical Practice Guidelines in Oncology: Multiple Myeloma version 4.2015. Available at http://www.nccn.org/professionals/physician_gls/pdf/myeloma.pdf accessed 23 April 2015.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015; 372: 142–152.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hajek R et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016; 17: 27–38.
Acknowledgements
The study was funded by Onyx Pharmaceuticals, Inc., an Amgen subsidiary. We wish to thank all of the patients, families, caregivers, research nurses, study coordinators and support staff who contributed to this study. Medical writing assistance was provided by Christopher M. Brown, PhD, of BlueMomentum, an Ashfield Company, and funded by Onyx Pharmaceuticals, Inc., an Amgen subsidiary.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Hájek reports funding from Celgene, Janssen and Merck outside of the submitted work; Dr Masszi has nothing to disclose; Dr Petrucci reports receiving personal fees from Janssen-Cilag, Celgene, and BMS outside the submitted work; Dr Palumbo reports receiving personal fees from Amgen, Bristol-Myers Squibb, Genmab A/S, Janssen-Cilag, Millennium Pharmaceuticals, Onyx Pharmaceuticals, Sanofi Aventis, Celgene and Array BioPharma outside the submitted work; Dr Rosiñol reports personal fees from Onyx during the conduct of the study, and personal fees from Janssen, and Celgene outside the submitted work; Dr Nagler reports funding from Onyx outside the submitted work; Dr Yong has nothing to disclose; Dr Oriol reports other from Celgene, Jansen and Novartis outside the submitted work; Dr Minarik has nothing to disclose; Dr Pour has nothing to disclose; Dr Dimopoulos reports personal fees from Onyx and Celgene outside the submitted work; Dr Maisnar reports funding from Celgene, Janssen, and Amgen outside the submitted work; Dr Rossi has nothing to disclose; Dr Kasparu has nothing to disclose; Dr Droogenbroeck has nothing to disclose; Dr Ben-Yehuda has nothing to disclose; Dr Hardan has nothing to disclose; Dr Jenner received honoraria from Amgen/Onyx outside the submitted work; Dr Calbecka has nothing to disclose; Dr David has nothing to disclose; Dr de la Rubia has nothing to disclose; Dr Drach has nothing to disclose; Dr Gasztonyi has nothing to disclose; Dr Górnik has nothing to disclose; Dr Leleu reports funding from Celgene, Janssen, Leopharma, Amgen, Novartis, and Sanofi outside the submitted work; Dr Munder has nothing to disclose; Dr Offidani reports other from Celgene, Janssen, Amgen, Novartis, and Mundipharma outside the submitted work; Dr Zojer received honoraria from Janssen-Cilag and Mundipharma; Dr Chang is employed by, and owns stock in, Onyx; Dr Rajangam is employed by, and owns stock in, Onyx; Dr San Miguel reports funding from Millennium, Celgene, Novartis, Onyx, Janssen, BMS, and MSD outside the submitted work; and Dr Ludwig reports grants from Takeda, personal fees from Janssen-Cilag, Celgene, Bristol Meyers, Amgen and Onyx outside the submitted work.
Competing interests
The authors declare no conflict of interest.
Additional information
Results of this clinical trial were previously presented at the 2014 ESMO Annual Meeting (Ludwig et al. Ann Oncol 2014; 25(Suppl 4): LBA28).
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hájek, R., Masszi, T., Petrucci, M. et al. A randomized phase III study of carfilzomib vs low-dose corticosteroids with optional cyclophosphamide in relapsed and refractory multiple myeloma (FOCUS). Leukemia 31, 107–114 (2017). https://doi.org/10.1038/leu.2016.176
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.176
This article is cited by
-
Carfilzomib-related glomerular and tubular injury in a patient with Multiple Myeloma
Journal of Nephrology (2022)
-
Infection risks in multiple myeloma: a systematic review and meta-analysis of randomized trials from 2015 to 2019
BMC Cancer (2021)
-
A study of carfilzomib and dexamethasone in patients with relapsed and refractory multiple myeloma in China
International Journal of Hematology (2021)
-
A dose-finding Phase 2 study of single agent isatuximab (anti-CD38 mAb) in relapsed/refractory multiple myeloma
Leukemia (2020)
-
Risk of kidney toxicity with carfilzomib in multiple myeloma: a meta-analysis of randomized controlled trials
Annals of Hematology (2020)
