
Abstract
Hematopoietic stem/progenitor cells (HSPCs) reside in the bone marrow (BM) microenvironment and are retained there by the interaction of membrane lipid raft-associated receptors, such as the α-chemokine receptor CXCR4 and the α4β1-integrin (VLA-4, very late antigen 4 receptor) receptor, with their respective specific ligands, stromal-derived factor 1 and vascular cell adhesion molecule 1, expressed in BM stem cell niches. The integrity of the lipid rafts containing these receptors is maintained by the glycolipid glycosylphosphatidylinositol anchor (GPI-A). It has been reported that a cleavage fragment of the fifth component of the activated complement cascade, C5a, has an important role in mobilizing HSPCs into the peripheral blood (PB) by (i) inducing degranulation of BM-residing granulocytes and (ii) promoting their egress from the BM into the PB so that they permeabilize the endothelial barrier for subsequent egress of HSPCs. We report here that hematopoietic cell-specific phospholipase C-β2 (PLC-β2) has a crucial role in pharmacological mobilization of HSPCs. On the one hand, when released during degranulation of granulocytes, it digests GPI-A, thereby disrupting membrane lipid rafts and impairing retention of HSPCs in BM niches. On the other hand, it is an intracellular enzyme required for degranulation of granulocytes and their egress from BM. In support of this dual role, we demonstrate that PLC-β2-knockout mice are poor mobilizers and provide, for the first time, evidence for the involvement of this lipolytic enzyme in the mobilization of HSPCs.
Similar content being viewed by others


Introduction
Hematopoietic stem/progenitor cells (HSPCs) express the chemokine receptor CXCR4 and the very late antigen 4 receptor (VLA-4, also known as α4β1-integrin) on their cell surface and are retained in the bone marrow (BM) niches by interaction of these receptors with their respective ligands, the α-chemokine stromal cell-derived factor 1 (SDF-1) and vascular adhesion molecule 1 (VCAM-1, also known as CD106), expressed by cells in the BM microenvironment (e.g., by osteoblasts and fibroblasts).1, 2, 3, 4, 5, 6 Mobilization studies using small-molecule antagonists of CXCR4 or VLA-4 indicate the importance of both axes in retention of HSPCs in the BM microenvironment.1, 2, 3, 4, 5, 6, 7 Moreover, evidence has accumulated that both receptors need to be incorporated into membrane lipid rafts for optimal function6, 7, 8, 9, 10, 11 and that a glycolipid, glycosylphosphatidylinositol anchor (GPI-A), has a pivotal role in maintaining the integrity of cell-surface membrane lipid rafts.6, 8, 11, 12 In support of this notion, patients suffering from paroxysmal nocturnal hemoglobinuria, in which HSPCs do not express GPI-A on the cell surface, show a profound defect in HSPC retention in BM niches.6, 8
GPI-A can also be removed from cell membranes by exposure to the lipolytic enzyme phospholipase C (PLC), which, if released from the cells, affects GPI-A by enzymatic digestion and thus disrupts lipid raft integrity.6 On the one hand, PLC is an intercellular enzyme that is released from cells during inflammation,13, 14, 15 and thus may affect cell-surface expression of proteins that are closely associated with GPI-A, including a truncated form of VCAM-1, the complement inhibitors CD55 and CD59, and uPAR.6, 16, 17 On the other hand, as an intracellular enzyme, PLC is involved in the chemotactic response of leukocytes to the cleavage fragment of the fifth protein component of the complement cascade (C5a).18, 19 Overall, the PLC family of enzymes consists of 13 members split between six subfamilies, including the PLC-δ (1, 3, 4), -β (1–4), -γ (1, 2), -ɛ, -ζ and -η (1, 2) isoforms. Among these isoforms, PLC-β2 is unique in being predominantly expressed in hematopoietic-specific enzyme.18, 19, 20
HSPCs are mobilized from BM niches into the peripheral blood (PB) after administration of the cytokine granulocyte colony-stimulating factor (G-CSF) or the small-molecule antagonist of the CXCR4 receptor AMD3100.1, 2, 3, 4, 5, 6, 21, 22, 23 Administration of these drugs activates several pathways in the BM microenvironment, including the complement cascade. In particular, activation of the distal part of the complement cascade releases C5a, which is processed to its long-lasting derivative desArgC5a. C5-deficient mice are poor mobilizers, which supports an important role for C5a and desArgC5a in the egress of HSPCs from the BM into the PB.24 The explanation for this finding is that both C5 cleavage fragments induce degranulation of BM-residing granulocytes, which releases proteolytic enzymes and promotes egress of these cells from the BM into the PB. These cells also permeabilize the endothelial barrier for subsequent egress of HSPCs.24, 25 As mentioned above, C5a interaction with the C5a receptor on granulocytes activates intracellular PLC-β2-mediated signaling.18, 19
Based on the potential dual role of PLC-β2 in the mobilization process, including (i) its potential extracellular effect as an enzyme that targets GPI-A and impairs lipid raft integrity and (ii) its role in intracellular C5a-mediated signal transduction, which leads to degranulation of granulocytes, we performed mobilization studies in PLC-β2-knockout (PLC-β2-KO) mice.
We demonstrate here for the first time that PLC-β2-deficient mice are poor mobilizers and that this lipolytic enzyme is involved in the mobilization process by (i) disrupting lipid raft integrity and (ii) enabling C5a-mediated release of proteolytic enzymes by granulocytes as well as their chemotactic migration in response to a C5a gradient between BM and PB.
Materials and methods
Animals
Six-week-old C57BL/6J wild-type mice (WT) and B6.129S1-Plcβ2tm1Dwu/J PLC-β2-KO mice of both sexes were used in our experiments. Mice from the Jackson Laboratory (Bar Harbor, ME, USA) were randomly divided into two groups of each sex. Animal studies were approved by the Animal Care and Use Committee of the University of Louisville (Louisville, KY, USA).
Isolation of Gr-1+ cells
Gr-1+ cells were isolated from the BM of adult male or female mice. Briefly, the BM was flushed from femurs, and the population of total nucleated cells was obtained after lysis of red blood cells (RBCs) using 1 × BD Pharm Lyse buffer (BD Pharmingen, San Jose, CA, USA). Cells were subsequently stained with phycoerythrin (PE)-anti-Gr-1 antibody (anti-Ly-6G and Ly-6C, clone RB6-8C5) or with PE-anti-CD11b antibody (clone M1/70) for 30 min in medium containing 2% fetal bovine serum. Cells were then washed, resuspended in RPMI-1640 medium and sorted using a Moflo XDP cell sorter (Beckman Coulter, Indianapolis, IN, USA) as populations of granulocytes (SSChighGr-1+) or monocytes (SSClowCD11b+).26, 27
Measuring PLC activity
Gr-1+ cells were resuspended in medium RPMI-1640 plus 0.5% bovine serum albumin (BSA, 2 ml cells per 400 μl medium) and incubated overnight at 37 °C. Subsequently, cells were stimulated by adding G-CSF (100 ng/ml), AMD3100 (3 μm), C5a (1 μm) or desArgC5a (140 ng/ml) and incubated for 6 h at 37 °C. The cells were centrifuged, and conditioned media (CM) were collected and diluted (1:5). PLC activity was measured using the Amplex Red Phospholipase C Assay Kit (A12218; Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol, by measuring the fluorescence in a fluorescence microplate reader (Beckman Coulter DTX 880 Multimode Detector, Beckman Coulter) using excitation in the range of 530–560 nm and emission detection at ~590 nm.
FACS analysis of GPI-anchored proteins
The enzymatic action of phosphatidylinositol-specific phospholipase C (Sigma-Aldrich, St Louis, MO, USA) results in the release of GPI-anchored proteins, such as VCAM-1, uPAR, CD55 and CD59, from the cell membranes. BM stromal cells and BM mononuclear cells (MNCs) were incubated with phosphatidylinositol-specific phospholipase C (5 or 100 mU/ml) for 3 h at 37 °C. Alexa Fluor 647-conjugated anti-mouse CD106 clone 429 (BioLegend, San Diego, CA, USA), PE-conjugated anti-mouse uPAR clone 109801 (R&D Systems, Minneapolis, MN, USA), PE-conjugated hamster anti-mouse CD55 and anti-mouse CD59a clone 7A6 (BD Biosciences, San Jose, CA, USA) were applied to detect GPI-anchored proteins.
Lipid raft detection
Murine tibias and femurs were flushed, and BM-derived MNCs were separated via Ficoll-Paque. Murine CD34–LSK (Lin–Sca-1+c-Kit+CD34–) cells were purified by using fluorescence-activated cell sorting (FACS) from BM-derived MNCs using the following lineage marker-specific antibodies: PE-anti-TCR γδ, clone GL3; PE-anti-CD11b, clone M1/70; PE-anti-TCR β-chain, clone H57-597; PE-anti-CD45R/B220, clone RA3-6B2; PE-anti-TER-119/erythroid cells, clone TER-119; PE-anti-Ly-6G and Ly-6C, clone RB6-8C5; PE-Cy5-anti-Ly-6A/E (Sca-1), clone E13-161.7; fluorescein isothiocyanate (FITC)-anti-CD117 (c-Kit), clone 2B8; biotin-anti-CD34, clone HM34; and APC-streptavidin). Human CD34+ cells were obtained from cord blood after Ficoll-Paque separation, and subsequent isolation of CD34+ cells was performed using the CD34 MicroBead Kit, human (Miltenyi Biotec, San Diego, CA, USA). Highly purified human HSC (Lin–CD38–CD45RA–CD34+CD90+CD49f+) and MPP (Lin–CD38–CD45RA–CD34+CD90–CD49f–) fractions were isolated by FACS28 using the following lineage marker-specific antibodies: FITC-anti-CD235a, clone GA-R2 (HIR2); FITC-anti-CD2, clone RPA-2.10; FITC-anti-CD3, clone UCHT1; FITC-anti-CD14, clone M5E2; FITC-anti-CD16, clone 3G8; FITC-anti-CD19, clone, HIB19; FITC-anti-CD24, clone ML5; FITC-anti-CD56, clone NCAM16.2; FITC-anti-CD66b, clone G10F5; PerCP-anti-CD38, clone HB-7; PE-anti-CD90 (Thy1), clone 5E10; PE-Cy7-anti-CD49f, clone GoH3; APC-Cy7-anti-CD45RA, clone HI100; and APC-anti-CD34, clone 581/CD34. Briefly, cells were plated on poly-l-lysine-coated plates overnight, then incubated with phosphatidylinositol-specific phospholipase C (Sigma-Aldrich) for 1 h, washed and fixed in 3.7% paraformaldehyde. The cholera toxin B subunit conjugated with FITC (Sigma-Aldrich) was applied to detect the ganglioside GM1, and mouse monoclonal anti-hCXCR4 IgG antibody (R&D Systems) and Alexa Fluor 594 goat anti-mouse IgG antibody (Invitrogen) were applied to detect CXCR4. VLA-4 was stained with monoclonal rat anti-mouse integrin α4 (CD49d) antibody (EMD Millipore, Billerica, MA, USA) and Alexa Fluor 594. Stained cells were examined, and images were generated using a FluoView FV1000 laser-scanning confocal microscope (Olympus America Inc., Center Valley, PA, USA).8
Transwell migration assay
BM-derived Gr-1+ cells from WT and PLC-β2-KO mice were resuspended in assay medium (RPMI-1640 plus 0.5% BSA). Assay medium (650 μl) alone and containing C5a (140 ng/ml) or desArgC5a (140 ng/ml) purchased from Calbiochem (La Jolla, CA, USA) was added to the lower chambers of a Costar Transwell 24-well plate (Corning Costar, Cambridge, MA, USA). Aliquots of cell suspension (1 × 106 cells per 100 μl) were loaded onto the upper chambers with 5 μm pore filters, and then incubated for 3 h (37 °C, 5% CO2). Cells from the lower chambers were harvested and scored by FACS analysis.24, 29
Signal-transduction studies
Sorted Gr-1+ cells from WT and PLC-β2-KO mice were incubated overnight at 37 °C to induce quiescence. Next, the cells were stimulated with either RPMI-1640 plus 0.5% BSA alone or with the complement promobilizing factors C5a (140 ng/ml) or desArgC5a (140 ng/ml) for 2 min at 37 °C and then lysed using RIPA lysis buffer supplemented with protease and phosphatase inhibitors (Santa Cruz Biotech, Dallas, TX, USA). The extracted proteins were then separated on a 4–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, and the fractionated proteins were transferred to a polyvinylidene fluoride membrane. Phosphorylated intracellular kinase p42/44 mitogen-activated protein kinase (phospho-p42/44 MAPK) and phospho-AKT were detected with phospho-specific p42/44 MAPK (clone 9101) and phospho-specific AKT (Ser473; clone 9271) rabbit polyclonal antibodies (Cell Signaling, Danvers, MA, USA), respectively. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG was used as a secondary antibody (Santa Cruz Biotech). To evaluate protein loading, blots were subjected to stripping using stripping buffer (Thermo Scientific, Waltham, MA, USA) and then reprobing with appropriate anti-rabbit p42/44 MAPK (clone 9102) and AKT (clone 9272) monoclonal antibodies (both from Cell Signaling). All membranes were treated with an enhanced chemiluminescence (ECL) reagent (Amersham Life Science, Little Chalfront, UK), dried and subsequently exposed to film (Hyperfilm, Amersham Life Sciences). For band visualization, an automatic film processor supplied with fresh, warm developer and fixer solutions was used.24, 29
Degranulation assay
Gr-1+ cells from WT and PLC-β2-KO mice were resuspended in medium RPMI-1640 plus 0.5% BSA (2 ml cells per 400 μl medium) and incubated overnight at 37 °C. Subsequently, cells were stimulated by adding C5a (140 ng/ml), desArgC5a (140 ng/ml) or medium alone as control and then incubated for 6 h at 37 °C. Cells were centrifuged, and CM were collected. Myeloperoxidase (MPO) activity was determined using 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) as described.30 Briefly, 20 μl samples were combined with 100 μl of 3,3′,5,5′-tetramethylbenzidine substrate solution, and the plate was incubated at 37 °C for 20 min. The reaction was stopped by adding 50 μl of 1 m H2SO4, and absorption was measured at 450 nm to estimate myeloperoxidase activity. Elastase activity was measured using the EnzChek Elastase Assay Kit, according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA, USA). Briefly, 100 μl fresh sample was incubated with 100 μl substrate solution (with 25 μg/ml DQ elastin) for up to 4 h at room temperature in the dark, and the resulting fluorescence was recorded at 515 nm emission following 505 nm excitation. All activity assays were performed in triplicate in 96-well microtiter plates and analyzed with a Beckman Coulter DTX 880 Multimode Detector. Results are shown as a percentage of control samples, P⩽0.05.
In vivo mobilization studies
Mice (WT and PLC-β2-KO) were mobilized with G-CSF (short incubation, 3 days; long incubation, 6 days; both 100 μg/kg per day, subcutaneous injection) and AMD3100 (1 day, 5 mg/kg, subcutaneous injection). At 6 h after the last G-CSF injection or 1 h after AMD3100 injection, the mice were bled from the retro-orbital plexus for plasma and hematology analysis, and PB was obtained from the vena cava (with a 25-gauge needle and 1 ml syringe containing 250 U heparin). MNCs were obtained by hypotonic lysis of RBCs in BD Pharm Lyse buffer (BD Biosciences) as described.31, 32 PLC activity in mouse mobilized plasma and CM derived from BM after AMD3100 and G-CSF injection was evaluated. Plasma and CM were diluted 1:500 and 1:5, respectively. A phospholipase C assay was performed according to the Amplex Red Phospholipase C Assay Kit protocol.
Generation of radiation chimeras
Twenty-four hours after lethal irradiation (1000 cGy), WT and PLC-β2-KO female mice were transplanted through the retro-orbital plexus with 5 × 106 BM MNCs from PLC-β2-KO or WT mice. Animals that had undergone transplantation were allowed to recover for 5 weeks, after which chimeras were mobilized and analyzed in the same way as described above for in vivo mobilization studies.
PB parameter counts
To obtain leukocyte and RBCs counts, 50 μl of PB was taken from the retro-orbital plexus of the mice into microvette EDTA-coated tubes (Sarstedt Inc., Newton, NC, USA) and run within 2 h of collection on a HemaVet 950FS hematology analyzer (Drew Scientific Inc., Oxford, CT, USA).24, 31
FACS analysis
The following monoclonal antibodies were used to perform staining of Lin–/Sca-1+/c-Kit+ (SKL) cells and Lin–/Sca-1+/CD45+ (HSCs): FITC–anti-CD117 (also known as c-Kit, clone 2B8; BioLegend) and PE-Cy5-anti-mouse Ly-6 A/E (also known as Sca-1, clone D7; eBioscience, San Diego, CA, USA). All anti-mouse lineage markers (Lin) were purchased from BD Biosciences: anti-CD45R/B220 (clone RA3-6B2), anti-Ter-119 (clone TER-119), anti-CD11b (clone M1/70), anti-T-cell receptor β (clone H57-597), anti-Gr-1 (clone RB6-8C5), anti-TCRγδ (clone GL3) and anti-CD45 (clone 30-F11) and conjugated with PE as described.24, 29, 31, 32 All monoclonal antibodies were added at saturating concentrations, and the cells were then incubated for 30 min on ice, washed twice, resuspended in RPMI-1640 plus 2% fetal bovine serum, and analyzed with an LSR II flow cytometer (BD Biosciences, San Diego, CA, USA).
Evaluation of HSPC mobilization
For evaluation of circulating colony-forming unit-granulocyte/macrophage (CFU-GM) and SKL cells, the following formulas were used: (number of white blood cells (WBCs) × number of CFU-GM colonies)/number of WBCs plated=number of CFU-GM per ml of PB; and (number of WBCs × number of SKL cells)/number of gated WBCs=number of SKL cells per μl of PB.24, 29, 31, 32
Fibronectin cell adhesion assay
BM MNCs were made quiescent for 3 h with RPMI-1640 plus 0.5% BSA medium and incubation with phosphatidylinositol-specific phospholipase C (100 and 1 U/ml) for 10, 30 or 60 min. Subsequently, cell suspensions (15 × 104/100 μl) were added directly to 96-well plates coated with fibronectin (10 μg/ml) with or without SDF-1 (300 ng/ml) for 15 min at 37 °C. The wells were then washed of non-adherent cells, and all adherent cells were counted using an inverted microscope.8
Clonogenic in vitro assay
After RBCs from PB were lysed (Pharm Lyse buffer; BD Biosciences), nucleated cells from PB were subsequently washed two times and used for a CFU-GM assay. The cells were counted and resuspended in human methylcellulose base media provided by the manufacturer (R&D Systems), supplemented with 25 ng/ml recombinant murine granulocyte macrophage colony-stimulating factor (mGM-CSF) and 10 ng/ml recombinant murine interleukin-3 (Millipore, Billerica, MA, USA). Cultures were incubated for 7 days and then scored for the number of CFU-GM colonies under an inverted microscope. We cultured 1 × 106 PB MNCs per dish, and the final results were recalculated based on the number of PB MNCs per 1 μl of PB. Each clonogenic test was performed in duplicate. To evaluate the number of clonogenic progenitor cells, BM MNCs were supplemented with erythropoietin (5 U/ml; Stem Cell Tech., Vancouver, BC, Canada) plus stem cell factor (5 ng/ml) and resuspended in methylcellulose base medium (for determining the number of burst-forming unit-erythroid), supplemented with thrombopoietin (100 ng/ml) plus murine interleukin-3 (10 ng/ml), and the resuspended plasma clots used for determining the number of CFU-megakaryocytes. The CFU-GM assay was performed as above. Cultures were incubated for 7 days (37 °C, 95% humidity and 5% CO2), at which time they were scored under an inverted microscope for the number of each type of colony as described.29
Statistical analysis
All results are presented as mean±s.d. Statistical analysis of the data was carried out using Student's t-test for unpaired samples, with P⩽0.05 considered significant.
Results
PLC becomes activated in BM during HSPC mobilization
It has been reported that PLC is released from HSPCs during inflammation and that its extracellular concentration increases in body fluids.13, 14, 15 As mobilization of HSPCs is part of the innate immunity inflammatory response,4 we became interested in whether PLC is released from cells in response to G-CSF- or AMD3100-induced mobilization. To address this question, we measured PLC activity in blood plasma and in CM from cells recovered from the BM cavities of mobilized animals and observed an increase in PLC activity (Figure 1a).

PLC activation in response to promobilizing factors. (a) PLC activity was measured in plasma (left panel) and BM cell CM (right panel) from the BM of WT male and female mice mobilized with G-CSF or AMD3100. (b) PLC activity in CM from Gr-1+ cells sorted from female and male WT mice. BM was incubated for 6 h with G-CSF, AMD3100, C5a or desArgC5a. (c) Dose–response effect of promobilizing factors on PLC activation. PLC activity in CM from granulocyte and monocyte cells sorted from BM. BM-isolated granulocytes or macrophages were incubated for 6 h with G-CSF (10, 50, 100 and 500 ng/ml, left panel) or AMD3100 (0.3, 1, 3, 10 and 30 μm, right panel). The assay was performed according to the Amplex Red Phospholipase C Assay Kit protocol. Data from two separate experiments are pooled together and compared with untreated controls. *P⩽0.05 and **P⩽0.005.
Next, based on evidence that C5a activates granulocytes in a PLC-dependent manner, we sorted the BM Gr-1+ cells and stimulated them with both C5 cleavage fragments, C5a and desArgC5a, as well as with G-CSF and AMD3100. Figure 1b demonstrates that, upon stimulation, BM-purified Gr-1+ cells isolated from BM secrete PLC into the culture medium. Interestingly, we also observed some differences in the PLC level detected in preparations of Gr-1+ cells isolated from male and female mice. As shown in Figure 1b, female mice secreted more PLC-β2 than males upon stimulation.
Finally, we performed dose–response studies on granulocytes (SSChighGr-1+ cells) and monocytes (SSClowCD11b+ cells) purified from murine BM.26, 27 Cells were stimulated (Figure 1c) with increasing doses of G-CSF (left panel) or AMD3100 (right panel), and PLC-β2 was detected by enzyme-linked immunosorbent assay. It can be seen that PLC-β2 activity was detected in CM harvested from granulocytes.
PLC targets several molecules involved in retention of HSPCs in the BM
Based on the finding that PLC is released during mobilization from granulocytes (Figure 1a), we focused on potential GPI-A+ targets for this enzyme, including the murine short isoform of VCAM-1, uPAR and the complement cascade inhibitors CD59 and CD55.6, 16 As shown in Figure 2a, exposure of BM-derived murine mesenchymal stromal cells to PLC resulted in dose-dependent removal from the cell membrane of the GPI-A+ truncated form of VCAM-1 and uPAR (Figure 2a, left panel). Similarly, exposure of BM MNCs to PLC led to a decrease in the expression of GPI-A+ cell-surface complement inhibitors CD59 and CD55 (Figure 2a, right panel).

Impact of PLC on GPI-A+ proteins expressed on BM cells. (a) Expression of VCAM-1 and uPAR receptors (left panel) on BM MNCs after PLC treatment (5–100 mU/ml) and cell-surface expression of membrane complement regulators CD55 and CD59 on BM MNCs (right panel) after PLC treatment (100 mU/ml). Data from three separate experiments are pooled together and compared with untreated controls. *P⩽0.05 and **P⩽0.005. (b) Defective lipid raft formation in murine BM-purified CD34–LSK cells. Representative images of CD34–LSK cells sorted from BM, stimulated by LL-37 (2.5 μg/ml), stained with cholera toxin subunit B (a lipid raft marker) conjugated with rat anti-mouse VLA-4 and Alexa Fluor 594, and evaluated by confocal microscopy for formation of membrane lipid rafts. Lipid rafts were formed in CD34–LSK cells (control), but not in CD34–LSK cells after PLC treatment.
Moreover, as the glycolipid GPI-A is required for the proper function of lipid rafts,6, 8, 11, 12 we exposed murine CD34–LSK cells enriched for HSPCs to PLC and evaluated the effect on lipid raft integrity (Figure 2b). We found that murine CD34–LSK cells exposed to PLC display defective lipid raft formation, which is crucial for CXCR4- and VLA-4-mediated retention of HSPCs in the BM.1, 2, 3, 4, 5, 6, 7 Similar results were obtained with human Lin–CD38–CD45RA–CD34+CD90–CD49f– cells (Supplementary Figure 1). Furthermore, our adhesion experiments demonstrated that, if exposed to PLC, murine Sca-1+ cells show defective adhesion to mesenchymal stromas or immobilized SDF-1 (Supplementary Figure 2).
This part of our work indicates that, as an extracellular enzyme released pharmacologically to induce mobilization, PLC perturbs the integrity of membrane lipid rafts and thus may affect retention of HSPCs in their BM niches.
Defect in C5a- and desArgC5a-mediated degranulation and chemotactic response in PLC-β2 granulocytes
Based on reports that hematopoietic cell-specific PLC (PLC-β2) is involved in C5a signaling,18, 19 we became interested in the role of PLC-β2 in C5a- and desArgC5a-mediated responses of murine BM-derived Gr-1+ leukocytes. These cells are the first to be mobilized into the PB and have a role in permeabilization of the endothelial–BM barrier to pave the way for subsequent egress of HSPCs.21, 24, 25
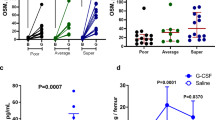
To address this issue, we evaluated the chemotactic responsiveness to C5a and desArgC5a gradients of murine BM-derived Gr-1+ cells isolated from the BM of WT and PLC-β2-KO mice (Figure 3a). As shown, we found impaired chemotactic responsiveness of these cells in a Transwell migration assay, which was supported by signal-transduction studies in which we observed a severe reduction in MAPK p42/44 phosphorylation in Gr-1+ cells from PLC-β2-KO animals (Figure 3b). This result was corroborated by the results of a degranulation assay performed in Gr-1+ cells isolated from WT and PLC-β2-KO mice stimulated with C5a or desArgC5a (Figure 3c). We found that the release of myeloperoxidase and elastase from PLC-β2-KO Gr-1+ cells was significantly impaired.

Defective promobilizing properties of Gr-1+ cells isolated from PLC-β2-KO mice. (a) Chemotactic responsiveness of BM-derived Gr-1+ cells purified from WT and PLC-β2-KO mice to medium alone (RPMI-1640 plus 0.5% BSA), C5a (140 ng/ml) or desArgC5a (140 ng/ml). Results are combined from two independent experiments and shown as the number of cell events by FACS analysis. *P⩽0.05. (b) The effect of complement C5 cleavage fragments on phosphorylation of MAPK p42/44 and AKT Ser473 in Gr-1+ cells purified from WT and PLC-β2-KO mice. Cells (1 × 106 cells per ml) were initially starved overnight in RPMI medium containing 0.5% BSA at 37 °C and later stimulated in vitro by the C5 cleavage fragments C5a (140 ng/ml) or desArgC5a (140 ng/ml) for 2 min. Note that MAPK and AKT phosphorylation in response to either C5a or desArgC5a is significantly weaker in PLC-β2-KO mouse-derived cells compared with cells from WT animals. The experiment was carried out two times with similar results, and a representative blot is shown. (c) A defect in C5a- and desArgC5a-mediated granulocyte degranulation in cells from PLC-β2-KO mice. Degranulation of BM-derived Gr-1+ cells sorted from WT and PLC-β2-KO mice was measured based on MPO released from the cells during the degranulation process (left panel) and elastase (right panel) activity. Cells were incubated with medium alone (control), C5a (140 ng/ml) or desArgC5a (140 ng/ml) for 6 h. Subsequently, cells were centrifuged, and the CM collected and analyzed. The results are combined from three independent experiments and show changes as a percentage of control. *P⩽0.05.
Based on this finding, we conclude that PLC-β2 is involved as an intracellular enzyme in degranulation and release of proteolytic enzymes, as well as in normal migration and egress of granulocytes from the BM into the PB.
PLC-β2-deficient mice are poor G-CSF and AMD3100 mobilizers
Based on the above observations, we moved to a murine model of PLC-β2 deficiency and used PLC-β2-KO animals. Supplementary Figure 3 shows that PLC-β2-KO mice have normal PB cell counts (Supplementary Figure 3A), RBC parameters (Supplementary Figure 3B) and number of BM-residing HSCs (Supplementary Figure 3C) and clonogenic progenitors (Supplementary Figure 3D) compared with WT animals under steady-state conditions. At the same time, we did not observe any sex-dependent differences.
Next, we performed in vivo mobilization experiments. As we had observed some sex differences in the release of PLC during the mobilization process (Figure 1b), we used both female and male PLC-β2−/− animals in our experiments. Figure 4 shows that PLC-β2-KO mice had impaired day 3 G-CSF- (Figure 4a) and AMD3100-induced mobilization (Figure 4b). Moreover, despite differences in PLC release form Gr-1+ cells (Figure 1b), we did not observe any sex-related differences in egress of HSPCs from the BM into the PB. Similar results were obtained with PLC-β2-KO mice after G-CSF-induced mobilization was prolonged to 6 days (Supplementary Figure 4).

PLC-β2-KO mice are poor G-CSF and AMD3100 mobilizers. (a) MNCs were isolated from WT and PLC-β2-KO mice after G-CSF mobilization (for 3 days at 100 μg/kg per day, subcutaneously). Upper panel, results from female mice; lower panel, results from male mice. (b) MNCs were isolated from WT and PLC-β2-KO mice after AMD3100-induced mobilization (single dose at 5 μg/kg, intraperitoneal injection). Upper panel, data from female mice; lower panel, data from male mice. In both experiments, mice were killed 6 h after the last G-CSF injection and 1 h after AMD3100 mobilization, and the numbers of WBCs, SKL (Sca-1+ c-kit+ Lin−) cells, HSCs (Sca-1+ CD45+ Lin−) and CFU-GM clonogenic progenitors were evaluated from PB. Results from two separate experiments are pooled together, and results from female and male mice are shown separately. *P⩽0.05 and **P⩽0.005.
However, PLC-β2 has been reported to be hematopoietic cell-specific. To exclude the possibility that the defect observed in PLC-β2−/− mice on mobilization could also somehow be microenvironment-dependent, we created hematopoietic chimeras by transplanting BM cells from PLC-β2−/− mice into WT animals and vice versa. After chimeras were established, the mice were mobilized 5 weeks later by treatment with G-CSF or AMD3100. Figure 5 shows that WT chimeric mice transplanted with PLC-β2−/− BM cells showed defective short- and long-term mobilization induced by G-CSF (Figures 5a and b) and AMD3100 (Figure 5c). At the same time, no mobilization defect was observed in PLC-β2−/− mice reconstituted with WT BM cells. This result excludes any potential involvement of residual PLC-β2 activity in non-hematopoietic tissues.

Mobilization of radiation chimeras. (a and b) WT chimeric mice reconstituted with PLC-β2-KO BM and PLC-β2-KO chimeric mice transplanted with WT BM cells were mobilized with G-CSF for 3 days (a) or 6 days (b). (c) WT chimeric mice reconstituted with PLC-β2-KO BM and PLC-β2-KO chimeric mice transplanted with WT BM cells were mobilized with AMD3100. In both cases, mobilization was evaluated by the number of circulating WBCs, SKL cells and HSCs per microliter of PB by flow cytometry and in vitro CFU-GM clonogenic progenitors per milliliter of PB. *P⩽0.05 and **P⩽0.005.
Discussion
The salient observation of our study is that optimal mobilization of HSPCs requires activation of hematopoietic-specific PLC-β2. This lipolytic enzyme has an important role in this process by regulating (i) the intracellular promobilizing functions of Gr-1+ cells and (ii) extracellular functions involving enzymatic perturbation of membrane lipid raft integrity, which is required for proper function of the CXCR4 and VLA-4 receptors and BM retention of HSPCs in their BM niches. This dual mechanism was supported by showing that PLC-β2 mice are poor mobilizers, despite the fact that under steady-state conditions these animals have normal PB cell counts and normal BM hematopoiesis.19
The mechanism of pharmacologically induced mobilization of HSPCs as a means to obtain stem cells for hematopoietic transplants is still not fully understood. Several signaling pathways regulating adhesion and migration of stem cells and regulating BM hematopoietic niche integrity, including Rac-1 and Rac-2 GTP-ases,9, 10, 11 glycogen synthase kinase-3β,33 cAMP-induced phosphokinase-ζ34 and PGE2,35 have been proposed to promote retention of HSPCs in the BM and their mobilization. Similarly, different types of BM cells have been proposed to have a crucial role in this process, including osteoblasts, osteoclasts,1, 36, 37, 38 osteomacs,1, 38 nestin+ mesenchymal stromas,39 CXCL12-abundant reticular cells,40 granulocytes41 and monocytes.42
Moreover, while the proretention function of osteoblasts, CXCL12-abundant reticular cells or nestin+ mesenchymal stromas is acknowledged, the role of osteoclasts in mobilization of HSPCs is still somehow controversial.37, 43, 44 Furthermore, as reported, mice with knockout of multiple proteolytic enzymes, including matrix metalloproteinase-9, matrix metalloproteinase-2, cathepsin G and elastase, mobilize HSPCs in a similar manner as WT control littermates.45 This finding suggests that deficiency in these enzymes is compensated by others such as, for example, CD26 (also known as dipeptidylpeptidase-4). Another candidate enzyme is cathepsin K, which is expressed, for example, by osteoclasts,46 and direct mobilization studies in cathepsin K-KO mice could answer this question.
For several years our group has been investigating the role of innate immunity, including the complement cascade,4, 24, 47, 48 coagulation cascade and fibrinolytic cascade,31 in pharmacologically induced HSPC mobilization, as we envision that this process is related to the normal response of BM and the hematopoietic system to stress situations seen in inflammation or tissue/organ injuries.4, 36
Pharmacologically induced mobilization can be achieved after administration of G-CSF or the CXCR4 antagonist AMD3100.1, 2, 3, 4, 5, 6, 21, 22, 23 The first and most important step in this process is detachment of HSPCs from the stem cell niches in the BM, where they are anchored by the ligands SDF-1 and VCAM-1, which are expressed in these niches. These ligands interact with the CXCR4 and α4β1-integrin (VLA-4) receptors, respectively, expressed on the surface of HSPCs. Both CXCR4 and VLA-4 are cell membrane lipid raft-associated receptors, and their presence in lipid rafts is essential to their optimal biological function.2, 6, 8, 9, 10, 11 Cell membrane lipid raft integrity is governed by GPI-A, which is a substrate for the lipolytic enzyme PLC.6, 8, 11, 16 It is widely accepted that an important role in pharmacological mobilization is played by induction of a proteolytic microenvironment in the BM because of the release of proteolytic enzymes from granulocytes and monocytes that digest proteins of the BM retention axes for HSPCs (e.g., SDF-1–CXCR4 and VCAM-1–VLA-4).49
Here, for the first time, we propose a novel mechanism in which, in addition to proteolytic enzymes, lipolytic enzymes such as PLC are released in the BM during the mobilization process, and a lipolytic microenvironment in the BM is created. The phenomenon of PLC release from cells during inflammation has been described in several pathological situations, such as inflammation, brain damage or cardiovascular complications,13, 14, 15, 50, 51 but so far it has not been recognized during pharmacologically induced mobilization of HSPCs. Based on the results reported in this paper, we have to consider the possibility that the mobilization process not only induces a proteolytic49 but also a lipolytic microenvironment in the BM. As demonstrated in our work, PLC released from Gr-1+ cells provides the basis for a novel mechanism promoting egress of HSPCs from the BM into the PB. This mechanism is based, on the one hand, on PLC-dependent digestion of GPI-A, leading to disintegration of lipid rafts, which results in perturbed CXCR4- and VLA-4-mediated retention of HSPCs in BM niches. An important role for lipid rafts in retention of HSPCs in the BM supports the concept of enhanced release of HSPCs into the PB in patients suffering from paroxysmal nocturnal hemoglobinura, a disorder in which HSPCs acquire a GPI-A mutation.6, 8, 16 Our results also explain in a novel way the increase in the soluble uPAR level that is observed in experimental and clinical mobilization.6, 17, 52 As uPAR is closely associated with a GPI-A fragment,6, 17 it becomes an easy substrate for PLC and thus is released from the cells.
Supporting evidence indicates that mobilizing agent-activated complement cascade and C5 cleavage fragments (C5a and desArgC5a) trigger egress of cells from the BM into the PB.6, 24, 25, 31, 32 To explain this observation, both of these molecules, also known as anaphylatoxins, are potent stimulators of degranulation and are chemoattractants of granulocytes and monocytes.6, 24, 25, 31, 32 We have proposed that mobilization-induced C5a/desArgC5a levels in PB chemoattractant granulocytes and monocytes, and egress of these cells from the BM into the PB facilitates subsequent egress of HSPCs.4, 24, 25
As reported previously, C5 deficiency,24 lack of granulocytes41 or a compromised function of monocytes42 all result in impaired egress of HSPCs from the BM into the PB. Our results reported in this paper support a promobilization link between C5a/desArgC5a signaling, PLC-β2 and granulocyte function. PLC-β2 is required for normal function of Gr-1+ cells, as we have shown that Gr-1+ cells from PLC-β2-KO mice respond very poorly to C5a/desArgC5a stimulation in degranulation assays and show impaired migration in response to C5 cleavage fragments. This defective responsiveness of Gr-1+ cells to C5a and desArgC5a is one of the reasons why PLC-β2-KO mice are poor mobilizers, as reported in our study.
Therefore, our results provide, for the first time, evidence that not only are proteolytic enzymes, such as MMP-9, MMP-2, cathepsin G, elastase, and CD26, activated during mobilization of HSPCs5, 45, 46, 49 but lipolytic enzymes such as PLC-β2 are also involved in this process, which was not previously appreciated. We envision that, in addition to PLC-β2, other enzymes also affect expression of the bioactive lipid sphingosine-1-phosphate (S1P). As recently demonstrated, mice with decreased activity of S1P kinase type 1 in BM, resulting in low BM expression of S1P, have a defect in retention of HSPCs in BM.53 Similarly, we expect that a high level of activity of the S1P-degrading enzyme S1P lyase has a similar effect. The importance of PLC-β2 and potentially other lipolytic enzymes in the retention of stem cells in BM may better explain why mice deficient in several proteolytic enzymes still mobilize HSPCs into the PB.45
Figure 6 summarizes our current understanding of the role of PLC-β2 in retention and egress of HSPCs. As demonstrated, intracellular PLC-β2 is involved in C5a/desArgC5a signaling-mediated degranulation of Gr-1+ cells and chemotactic responsiveness of these cells to a PB–BM gradient of both C5 cleavage fragments. This process then facilitates egress of HSPCs across the BM–PB endothelial barrier. PLC-β2 released from granulocytes also has an additional function as an extracellular enzyme that disrupts membrane lipid rafts and thus impairs retention of HSPCs in BM niches. All these biological effects of PLC-β2 together facilitate egress of cells into the PB.

Pleiotropic effects of PLC-β2 on the mobilization of HSPCs. (Right panel) Intracellular PLC-β2 is involved in C5a/desArgC5a signaling-mediated degranulation of Gr-1+ cells and chemotactic responsiveness of these cells to a C5 cleavage fragment gradient in the PB. This process of C5a/desArgC5a-mediated egress of granulocytes and monocytes from BM facilitates subsequent egress of HSPCs across the BM–PB endothelial barrier. (Left panel) PLC-β2 released from granulocytes during C5a/desArgC5a-mediated degranulation has an additional function as an extracellular enzyme that disrupts membrane lipid rafts by digesting GPI-A and thus impairing retention of HSPCs in BM niches. All these biological effects of PLC-β2 together facilitate egress of HSPCs into the PB.
In conclusion, we have established for the first time that, in addition to proteolytic enzymes, the lipolytic enzyme PLC-β2 is upregulated in the BM microenvironment during pharmacologically induced mobilization and promotes mobilization of HSPCs by inducing the promobilization function of Gr-1+ cells and perturbing BM retention of HSPCs. These results also support an important role for GPI-A-dependent proteins in the retention of HSPCs in BM niches and shed more light on C5a/desArgC5a-mediated egress of these cells from the BM into the PB in a PLC-β2-dependent manner.
References
Lévesque JP, Helwani FM, Winkler IG . The endosteal ‘osteoblastic' niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010; 24: 1979–1992.
Bonig H, Papayannopoulou T . Hematopoietic stem cell mobilization: updated conceptual renditions. Leukemia 2013; 27: 24–31.
Lapidot T, Kollet O . The brain–bone–blood triad: traffic lights for stem-cell homing and mobilization. Hematol Am Soc Hematol Educ Program 2010; 2010: 1–6.
Ratajczak MZ, Kim CH, Wojakowski W, Janowska-Wieczorek A, Kucia M, Ratajczak J . Innate immunity as orchestrator of stem cell mobilization. Leukemia 2010; 24: 1667–1675.
Doan PL, Chute JP . The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia 2012; 26: 54–62.
Ratajczak MZ, Adamiak M . Membrane lipid rafts, master regulators of hematopoietic stem cell retention in bone marrow and their trafficking. Leukemia 2015; 29: 1452–1457.
Lapidot T, Dar A, Kollet O . How do stem cells find their way home? Blood 2005; 106: 1901–1910.
Ratajczak MZ, Borkowska S, Mierzejewska K, Kucia M, Mendek-Czajkowska E, Suszynska M et al. Further evidence that paroxysmal nocturnal haemoglobinuria is a disorder of defective cell membrane lipid rafts. J Cell Mol Med 2015; 20: 1–9.
Chae HD, Lee KE, Williams DA, Gu Y . Cross-talk between RhoH and Rac1 in regulation of actin cytoskeleton and chemotaxis of hematopoietic progenitor cells. Blood 2008; 111: 2597–2605.
Wysoczynski M, Reca R, Ratajczak J, Kucia M, Shirvaikar N, Honczarenko M et al. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood 2005; 105: 40–48.
Capitano ML, Hangoc G, Cooper S, Broxmeyer HE . Mild heat treatment primes human CD34(+) cord blood cells for migration towards SDF-1α and enhances engraftment in an NSG mouse model. Stem Cell 2015; 33: 1975–1984.
Szpurka H, Schade AE, Jankowska AM, Maciejewski JP . Altered lipid raft composition and defective cell death signal transduction in glycosylphosphatidylinositol anchor-deficient PIG-A mutant cells. Br J Haematol 2008; 142: 413–422.
Meyers DJ, Berk RS . Characterization of phospholipase C from Pseudomonas aeruginosa as a potent inflammatory agent. Infect Immun 1990; 58: 659–666.
Jakus Z, Simon E, Frommhold D, Sperandio M, Mócsai A . Critical role of phospholipase Cγ2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med 2009; 206: 577–593.
König B, Jaeger KE, Sage AE, Vasil ML, König W . Role of Pseudomonas aeruginosa lipase in inflammatory mediator release from human inflammatory effector cells (platelets, granulocytes, and monocytes). Infect Immun 1996; 64: 3252–3258.
Brodsky RA . How do PIG-A mutant paroxysmal nocturnal hemoglobinuria stem cells achieve clonal dominance? Expert Rev Hematol 2009; 2: 353–356.
Stahl A, Mueller BM . The urokinase-type plasminogen activator receptor, a GPI-linked protein, is localized in caveolae. J Cell Biol 1995; 129: 335–344.
Buhl AM, Avdi N, Worthen GS, Johnson GL . Mapping of the C5a receptor signal transduction network in human neutrophils. Proc Natl Acad Sci USA 1994; 91: 9190–9194.
Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D . Roles of phospholipase C β2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA 1997; 94: 7971–7975.
Gresset A, Sondek J, Harden TK . The phospholipase C isozymes and their regulation. Subcell Biochem 2012; 58: 61–94.
Lee HM, Wysoczynski M, Liu R, Shin D-M, Kucia M, Botto M et al. Mobilization studies in complement-deficient mice reveal that optimal AMD3100 mobilization of hematopoietic stem cells depends on complement cascade activation by AMD3100-stimulated granulocytes. Leukemia 2010; 24: 573–582.
Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med 2005; 201: 1307–1318.
Flomenberg N, Devine SM, Dipersio JF, Liesveld JL, McCarty JM, Rowley SD et al. The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 2005; 106: 1867–1874.
Lee HM, Wu W, Wysoczynski M, Liu R, Zuba-Surma EK, Kucia M et al. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia 2009; 23: 2052–2062.
Ratajczak MZ . A novel view of the adult bone marrow stem cell hierarchy and stem cell trafficking. Leukemia 2015; 29: 776–782.
Zhang D, Jiang X, Fang P, Yan Y, Song J, Gupta S et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009; 120: 1893–1902.
Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol 2004; 172: 4410–4417.
Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE . Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011; 333: 218–221.
Ratajczak MZ, Lee HM, Wysoczynski M, Wan W, Marlicz W, Laughlin MJ et al. Novel insight into stem cell mobilization-plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia 2010; 24: 976–985.
Lacy P, Mahmudi-Azer S, Bablitz B, Hagen SC, Velazquez JR, Man SF et al. Rapid mobilization of intracellularly stored RANTES in response to interferon-γ in human eosinophils. Blood 1999; 94: 23–32.
Borkowska S, Suszynska M, Mierzejewska K, Ismail A, Budkowska M, Salata D et al. Novel evidence that crosstalk between the complement, coagulation, and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs). Leukemia 2014; 28: 2148–2154.
Wysoczynski M, Ratajczak J, Pedziwiatr D, Rokosh G, Bolli R, Ratajczak MZ . Identification of heme oxygenase 1 (HO-1) as a novel negative regulator of mobilization of hematopoietic stem/progenitor cells. Stem Cell Rev 2015; 11: 110–118.
Dolnikov A, Xu N, Shen S, Song E, Holmes T, Klamer G et al. GSK-3β inhibition promotes early engraftment of ex vivo-expanded haematopoietic stem cells. Cell Prolif 2014; 47: 113–123.
Goichberg P, Kalinkovich A, Borodovsky N, Tesio M, Petit I, Nagler A et al. cAMP-induced PKCzeta activation increases functional CXCR4 expression on human CD34+ hematopoietic progenitors. Blood 2006; 107: 870–879.
Hoggatt J, Pelus LM . Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia 2010; 24: 1993–2002.
Ratajczak MZ . Spotlight series on stem cell mobilization: many hands on the ball, but who is the quarterback? Leukemia 2010; 24: 1665–1666.
Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, To LB, Lévesque JP . Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood 1998; 92: 3465–3473.
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010; 116: 4815–4828.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 2011; 208: 261–271.
Sugiyama T, Kohara H, Noda M, Nagasawa T . Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006; 25: 977–988.
Pruijt JF, Verzaal P, van Os R, de Kruijf EJ, van Schie ML, Mantovani A et al. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin-8 in mice. Proc Natl Acad Sci USA 2002; 99: 6228–6233.
Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC . Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med 2011; 208: 251–260.
Rao M, Suparkorndej T, Schmidt AP, Link DC . Osteoclasts are dispensable for hematopoietic progenitor mobilization by granulocyte colony-stimulating factor in mice. Exp Hematol 2015; 43: 110–114.
Christopherson KW II, Hangoc G, Mantel CR, Broxmeyer HE . Modulation of hematopoietic stem cell homing and engraftment by CD26. Science 2004; 305: 1000–1003.
Lévesque JP, Liu F, Simmons PJ, Betsuyaku T, Senior RM, Pham C et al. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood 2004; 104: 65–72.
Kollet O, Dar A, Lapidot T . The multiple roles of osteoclasts in host defense: bone remodeling and hematopoietic stem cell mobilization. Annu Rev Immunol 2007; 25: 51–69.
Kim CH, Wu W, Wysoczynski M, Abdel-Latif A, Sunkara M, Morris A et al. Conditioning for hematopoietic transplantation activates the complement cascade and induces a proteolytic environment in bone marrow: a novel role for bioactive lipids and soluble C5b-C9 as homing factors. Leukemia 2012; 26: 106–116.
Ratajczak J, Reca R, Kucia M, Majka M, Allendorf DJ, Baran JT et al. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 2004; 103: 2071–2078.
Lévesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ . Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol 2002; 30: 440–449.
Nakashima T, Takenaka K, Nishimura Y, Andoh T, Sakai N, Yamada H et al. Phospholipase C activity in cerebrospinal fluid following subarachnoid hemorrhage related to brain damage. J Cereb Blood Flow Metab 1993; 13: 255–259.
Meij JT, Panagia V, Mesaeli N, Peachell JL, Afzal N, Dhalla NS . Identification of changes in cardiac phospholipase C activity in congestive heart failure. J Mol Cell Cardiol 1997; 29: 237–246.
Tjwa M, Sidenius N, Moura R, Jansen S, Theunissen K, Andolfo A et al. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. J Clin Invest 2009; 119: 1008–1018.
Adamiak M, Borkowska S, Wysoczynski M, Suszynska M, Kucia M, Rokosh G et al. Evidence for the involvement of sphingosine-1-phosphate in the homing and engraftment of hematopoietic stem cells to bone marrow. Oncotarget 2015; 6: 18 819–18 828.
Acknowledgements
This work was supported by NIH Grants 2R01 DK074720 and R01HL112788, the Stella and Henry Endowment and Maestro Grant 2011/0/A/NZ4/00035 (to MZR) and the UK COBRE Early Career Program (P20 GM103527), and NIH Grant R56 HL124266 to ABL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Adamiak, M., Poniewierska-Baran, A., Borkowska, S. et al. Evidence that a lipolytic enzyme—hematopoietic-specific phospholipase C-β2—promotes mobilization of hematopoietic stem cells by decreasing their lipid raft-mediated bone marrow retention and increasing the promobilizing effects of granulocytes. Leukemia 30, 919–928 (2016). https://doi.org/10.1038/leu.2015.315
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.315
This article is cited by
-
C3a and C5a facilitates the metastasis of myeloma cells by activating Nrf2
Cancer Gene Therapy (2021)
-
Innate immunity orchestrates the mobilization and homing of hematopoietic stem/progenitor cells by engaging purinergic signaling—an update
Purinergic Signalling (2020)
-
Pannexin-1 channel “fuels” by releasing ATP from bone marrow cells a state of sterile inflammation required for optimal mobilization and homing of hematopoietic stem cells
Purinergic Signalling (2020)
-
The Flot2 component of the lipid raft changes localization during neural differentiation of P19C6 cells
BMC Molecular and Cell Biology (2019)
-
NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow
Leukemia (2019)
