
Abstract
Genome-wide array approaches and sequencing analyses are powerful tools for identifying genetic aberrations in cancers, including leukemias and lymphomas. However, the clinical and biological significance of such aberrations and their subclonal distribution are poorly understood. Here, we present the first genome-wide array based study of pre-treatment and relapse samples from patients with B-cell chronic lymphocytic leukemia (B-CLL) that uses the computational statistical tool OncoSNP. We show that quantification of the proportion of copy number alterations (CNAs) and copy neutral loss of heterozygosity regions (cnLOHs) in each sample is feasible. Furthermore, we (i) reveal complex changes in the subclonal architecture of paired samples at relapse compared with pre-treatment, (ii) provide evidence supporting an association between increased genomic complexity and poor clinical outcome (iii) report previously undefined, recurrent CNA/cnLOH regions that expand or newly occur at relapse and therefore might harbor candidate driver genes of relapse and/or chemotherapy resistance. Our findings are likely to impact on future therapeutic strategies aimed towards selecting effective and individually tailored targeted therapies.
Similar content being viewed by others


Introduction
Genome-wide array approaches and sequencing analyses are powerful tools for identifying genetic aberrations in cancers, including leukemias and lymphomas.1, 2, 3, 4, 5, 6, 7, 8, 9, 10 B-cell chronic lymphocytic leukemia (B-CLL) is the most common adult leukemia in the Western World.11, 12 It is characterized by a chronic relapsing course and the development of chemotherapy refractoriness, leading to death in a significant subset of patients. There is, therefore, a clinical need to develop novel treatment strategies that overcome chemotherapy resistance.
B-CLL can be subclassified according to immunoglobulin heavy chain variable gene sequence (IGHV) homology into ‘mutated’ or ‘unmutated’ cases, reflecting good or poor prognosis, respectively. B-CLL shares many of the molecular characteristics seen in other forms of cancer and recurrent copy number alteration (CNAs) with relevance to prognosis have been described, including trisomy of chromosome 12 (16%) and loss of chromosomal regions 17p13.1 (TP53; 7%),13, 14, 15, 16, 17 11q22.3 (ATM; 18%)18, 19 and 13q14.3 (DLEU2 and/or miR-15a/16-1; 55%).20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 In addition, single-nucleotide polymorphism (SNP) and comparative genomic hybridization arrays have allowed novel recurrent genomic abnormalities to be identified31, 32, 33 that have been linked to prognosis in B-CLL.30, 34, 35, 36, 37, 38
Current models of cancer progression are based on the concept that tumors are subject to the Darwinian process of evolution and selection.39, 40 Recent studies in acute lymphoblastic leukemia have provided pivotal insights into the complex sequence of events during leukemogenesis, showing that the initiating mutation is followed by CNAs that drive the emergence of frank leukemia.39, 40, 41, 42 Together, these data imply that at least some CNAs/copy neutral loss of heterozygosity regions (cnLOHs) are likely to be involved in driving leukemia progression and therefore might contribute to relapse.
This led us to hypothesize that subclones containing driver CNAs/cnLOHs would newly occur or expand in relapse samples compared with samples taken before treatment and would be recurrent within our patient cohort. In contrast, random passenger mutations would remain unchanged or decrease/disappear in paired pre-treatment/relapse samples and would not be observed recurrently in different patients within the cohort. If correct, then we would anticipate that the identification of driver genes within recurrent or emerging/expanding regions of CNA/cnLOH might, in the longer term, have the potential to inform the design of novel therapies aimed at treating relapsed B-CLL.
In the present study we tested our hypothesis by systematically tracking the presence and subclonal distribution of CNAs/cnLOHs in patients before treatment and at subsequent relapse. To achieve this, we used high-resolution SNP array technology. We chose the newly developed computational statistical tool OncoSNP10 that provides quantitative measures of cell admixture on a per-SNP level. OncoSNP was selected in preference to other dedicated cancer tools for SNP array analysis,43, 44, 45, 46, 47 because our data were generated from samples at two time points only and our study was specific to B-CLL, where spatial heterogeneity (for example, samples biopsied from different parts of a tumor, metastases, etc) is not applicable (see Supplementary Information, online). Furthermore, we were interested not only in the accumulation of genomic aberrations over time, but also particularly in the expansion of distinct subclones. Thus, OncoSNP best suited our study where (i) samples differed from solid tumors in being generally diploid with comparatively fewer CNAs (ii) normal cell contamination could be minimized and (iii) we had a specific interest in subclonal evolution.
Patients and methods
For a detailed account of the Materials, Patients and Methods used in this study please refer to the Supplementary Information online.
Patients and samples
All patients gave written informed consent in accordance with the Declaration of Helsinki. In all, 135 samples from 93 patients were included in our cohort. For 42 of the 93 patients, paired pre-treatment and relapse samples were available. Only samples from patients with lymphocytes contributing >90% of the total white blood cell count were included in the analysis. DNA was extracted from vital frozen cells using the QIAamp DNA Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol.
Fluorescence in situ hybridization (FISH)
Interphase cells from CLL cultures were analyzed by FISH using the Vysis CLL FISH panel probe set (Abbott Molecular, Illinois, IL, USA) according to manufacturer's instructions.
Arrays
Hybridization to Illumina Genome-wide SNP chips was performed according to manufacturer's protocols found on registration at http://www.illumina.com/products/human1m_duo_dna_analysis_beadchip_kits.ilmn and at http://www.illumina.com/support/array/array_kits/humanomni1-quad_beadchip_kit/documentation.ilmn. The data were processed using GenomeStudioV2009.2 (Illumina, Inc., San Diego, CA, USA) and then analyzed using OncoSNP v1.0 (see below). For visual comparisons, the data were processed also using Nexus 5 Discovery Edition (BioDiscovery, Inc., El Segundo, CA, USA).
OncoSNP analysis
Detailed methods have been described previously10 and further details are given in the Supplementary Information online. To help identify and exclude germline CNVs of unlikely relevance from our data, we excluded SNP and monomorphic copy number probes in known germline CNV regions from the OncoSNP analysis and we also made use of the database of genomic variants48 and the Wellcome Trust Case Control Consortium cohort data (see www.wtccc.org.uk).
TP53 mutation analysis
TP53 was screened for mutations using four high-resolution melting assays covering exons 5–8. Following high-resolution melting, results were analyzed on the high-resolution melting module of the Corbett Rotor-Gene 6000 software 1.7 (available for download from QIAGEN, http://www.qiagen.com/corbett/support/default.aspx). Positive PCR products were purified and then sequenced from both strands on the CEQ 8000 (Beckman Coulter, Fullerton, CA, USA).
IGHV mutation analysis
To identify clonal rearrangements of the IGHV gene and determine the somatic mutation status of the variable (V) gene sequence in patients we used the IGH Somatic Hypermutation Assay v2.0 (Invivoscribe, La Coutat, France) according to the manufacturer's instructions.
Results
A flowchart outlining the patient numbers and overall study design is given in Supplementary Figure 1 online.
Our preliminary experiments demonstrated the importance of selecting an array platform capable of detecting CNAs and cnLOH (see Supplementary Information, Supplementary Table 1 and Figure 2 online) as we found regions of cnLOH signposting genes that might carry mutations relevant for treatment choice and that would otherwise have been missed using a non-SNP-based platform (for example, TP53, see Supplementary Information, Supplementary Table 2 and Figure 3 online). We also demonstrated in these experiments that OncoSNP provided accurate quantification of CNAs, based on the strong correlation between OncoSNP results and conventional FISH analysis (Supplementary Table 3 and Figure 4 online).
Genome-wide analysis of pre-treatment samples
For our main study, we focused initially on the analysis of pre-treatment samples from 93 patients undergoing regular follow-up at our institutions. This cohort was representative of CLL cohorts with a higher proportion of unmutated IGHV genes and/or treatment resistant cases that are referred to tertiary centers for treatment consideration (Table 1). A total of 80 patients had one or more CNAs/cnLOHs in well-established regions of prognostic importance - 17p13.1, trisomy 12, 13q14.3, 11q22.3 (Table 1, Supplementary Table 2, Figures 2, 3 and 5 online). CNAs involving chromosome arms 8p, 9p and 10q developed exclusively in patients with unmutated IGHV.
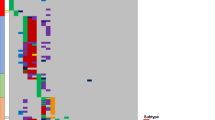
We identified 58 previously unreported recurrent CNAs/cnLOHs (Table 2) (see Supplementary Information online for recurrence criteria). We established the minimally deleted regions (MDRs)/minimally overlapping regions (MORs) of the recurrent CNAs/cnLOHs and revealed interesting candidate genes including those implicated in B-cell maturation (for example, BLIMP1, NFkB2, TLR4, and CREBBP), tumor progression (for example, RND3, RHOT1; RHDBL; RAB20, and TRAP1), DNA damage response (for example, RIF1, TP53, ATG4D, and ATG5), tumor suppression (for example, NMI, CNOT7, PDGFRL, FGF20, PI3 K, and FOG2) and familial CLL (for example, SP140L, SP100) (Table 2). Figure 1 shows examples of four of these novel recurrent regions of interest that span only a few candidate genes, potentially facilitating the identification of driver mutations. Sequencing of the TP53 gene, the known candidate underlying poor prognosis on 17p13.1, revealed mutations in 14 out of 93 patients and deletions in 11. However, exon sequencing of two of the emergent candidates, BLIMP1 and ATG5 revealed no mutations in 40 patients.

Four recurrent regions of interest observed in B-CLL patient samples. The boundaries of the MDRs/MORs, 2q37.1, 6q21, 9p22.3p24.1 and 10q24.32, are indicated by the vertical lines. Red lines show called losses, green lines show gains, purple lines show allelic imbalance and mustard lines show cnLOH. In each case the MDR/MOR was verified by visual inspection of Log ratio and B-allele frequency plots. P=Pre-treatment, R=Relapse.
In order to establish the clinical significance of our findings, we correlated our array results with measures of clinical prognosis and risk, including IGHV mutation status and clinical risk scores defined by necessity for treatment intervention, progression free survival and chemotherapy resistance (see Supplementary Information, online). Genomic complexity was defined either by the presence of three or more CNAs ⩾20 kb and/or cnLOHs ⩾2 Mb in addition to, or other than, the known CNA regions assayed routinely by FISH, or by a total length of CNAs ⩾5 Mb. Within the 93 pre-treatment samples, patients with unmutated IGHV genes showed a statistically significant higher total number of CNAs/cnLOHs (50.0% ⩾3 CNAs/cnLOHs) than patients with mutated IGHV genes (16.0% ⩾3 CNAs/cnLOHs) (Cochran–Armitage trend exact test, P-value=0.0032, Supplementary Table 4 online) and patients with del17p/TP53 mutation showed a higher total length of CNAs/cnLOHs (90.9% >5 Mb CNAs/cnLOHs) (Cochran–Armitage trend exact test, P-value=0.0081, Supplementary Table 5, online). In our small cohort, large Type II 13q deletions were not associated with a worse clinical outcome (Supplementary Tables 6 and 7 online).
Furthermore, patients with high clinical risk scores had a higher number of CNAs/cnLOHs than patients with low clinical risk scores (Kruskal–Wallis test, P-value=0.0016) and showed a greater overall length of CNAs/cnLOHs than patients with low clinical risk scores (Kruskal–Wallis test, P-value=0.0002) (Supplementary Table 8, online). Importantly, 16 patients had ⩾3 CNAs/cnLOH but no 17p13.1 or 11q22.3 loss and would not have been picked up by FISH as a poor risk group.
Genome-wide comparison of paired pre-treatment/relapse samples
Next, we focused on the analysis of the 42/93 patients for whom paired samples (that is, both pre-treatment and relapse samples) were available (Table 3). We hypothesized that in addition to recurrent CNAs, newly occurring or expanding genomic aberrations would also represent potential drivers of disease progression. The percentages of subclones carrying CNAs/cnLOH regions were calculated from the SNP data using OncoSNP.10 Only one case with mutated IGHV genes (CLL086) had additional CNAs at relapse. The other changes were percentage changes of pre-existing CNAs. By contrast, 11 unmutated IGHV genes cases had complex evolution with emergence of one or more additional CNAs. When we looked at total length of CNAs/cnLOHs, patients without clonal evolution showed a statistically significant lower total length of CNAs/cnLOHs (58.8%<1 Mb) than patients with clonal evolution (68.0% >5 Mb)(Cochran–Armitage trend exact test, P-value=0.0075; Supplementary Table 9, online).
All 13q14.3 deletions were present before treatment and of the 21 patients manifesting these; seven exhibited ⩾10% changed proportions of subclones carrying the deletion at relapse. In two cases, (CLL081 and CLL107), the proportion of subclones with the del13q14.3 appeared decreased at relapse and both exhibited additional CNAs elsewhere in the genome. For another two cases (CLL080 and CLL096), with deletions >1 Mb, the proportion of subclones with the 13q14.3 deletion had increased over time; both had isolated chromosome 13 anomalies (Figure 2). Furthermore, in CLL071 and CLL080, there was extension of the 13q13.4 deletion in one allele (Supplementary Figure 6 online, Supplementary Table 5). Overall, the results suggest that extension of the deleted 13q14.3 locus may confer a clonal survival advantage in the minority of cases without additional CNAs/cnLOHs. By contrast, for patients with complex genomic aberrations, clones containing del13q14.3 are outcompeted.

OncoSNP output showing clonal expansion events involving the chromosome 13q14.3 MDR and percentages of cells involved in pre-treatment and relapse samples from CLL080 and CLL096. The red lines indicate deletion events, whereas the magenta lines indicate allelic imbalance/cnLOH. In CLL080, 30% cells carry a large 13q14.11q14.3 deletion and an additional ∼20% cells carry the smaller MDR deletion at pre-treatment. At relapse, ∼90% cells carry the larger deletion. For CLL096, there is a homozygous deletion of 13q14.3, and cnLOH of the entire chromosome in ∼40% cells at pre-treatment. This cnLOH expands to ∼90% cells at relapse.
Patients with del17p13.1/TP53 mutations were most likely to acquire additional CNAs at relapse (5/5). In addition, three patients without del17p13.1 at diagnosis developed del17p and other CNAs at relapse (CLL084 and CLL108, CLL145). Patient CLL081 was a clear example of this, manifesting a 17p13.1 deletion and multiple other CNAs pre-treatment. At the time of relapse, five CNAs had altered in proportion (Figure 3) and two events (del2q33.1-q37.1 and del16p13) were newly identified. Furthermore, a number of the CNAs identified encompassed newly identified MDRs/MORs (Table 1) and included the genes LDB-1 and NFkB2 (10q24.32 MDR), SP140L and SP100 (2q37.1 MOR) and BTBD12, DNASE1, TRAP1 and CREBBP (16p13.3 MDR). Finally, the OncoSNP feature of defining proportions of subclones carrying CNAs/cnLOHs allowed us to use data from matched pre-treatment and relapse samples to infer likely subclonal populations and to map their evolutionary relationships. Schematic representations of the likely clonal architecture at different time-points for patients CLL081 and CLL106 are shown in Figure 4 with a further example for CLL092 in Supplementary Figure 7 online. These illustrate clearly a non-linear, branching subclonal hierarchy in B-CLL with multiple ancestral subclones, already present in pre-treatment samples and contributing to relapse. In conclusion, using the combination of high-resolution SNP array and OncoSNP, we were able to detect clonal evolution in 60% of treated patients with B-CLL.

OncoSNP output showing clonal evolution in pre-treatment and relapse samples from CLL081. This patient shows a reduction in the percentage of cells carrying the 13q abnormalities whilst other CNAs expanded (8q22.2qter and 10q23.2q23.3) or were newly identified at relapse (2q33.1q37.1 and 16p13.3).

Schematic representation of the possible clonal architecture pre-treatment and at relapse (pale blue and mid-blue shaded areas, respectively) for patients (a) CLL106 and (b) CLL081. Proportions below 10% and above 90% and cell populations without identifiable CNAs/cnLOHs are inferred. The precision of the percentage contribution of subclones with CNAs/cnLOH is ±10%. Note that for CLL081, the proportion of subclones with the 13q14.3 loss decreases at relapse and the 12q13.11q13.12 and 19p13.13p13.2 gains are no longer observed in one subclone.
Interestingly, when comparing the regions affected by clonal expansion with recurrent regions we noticed a considerable overlap (Table 3). Many regions that were both recurrent and expanded or newly occurred in relapse samples included a very limited number of genes (Table 2, blue shading). These regions include genes that have a role in familial CLL (SP140L, SP100), in B-cell development and autophagy (BLIMP1 and ATG5, respectively) and FOG2, a regulator of phosphatidylinositol 3-kinase, which is involved in cancer proliferation and survival. A fourth region affects NFIB involved in the NFKB pathway and two genes of no obvious CLL related annotation (TYRP1, MPDZ). A further region included DNA repair genes (BTBD12, DNASE1), TRAP1, a mitochondrial chaperone and regulator of apoptosis, as well as CREBBP, a frequently mutated gene in B-cell lymphomas.
When we performed pathway analysis of the 546 genes in the MDRs/MORs affected by recurrent and emerging/expanding CNAs, we identified 17 over-represented, statistically significant and independent pathways of which eight contained 55 cancer-related genes (phagosome P-value=0.0024; apoptosis P-value=0.0079; small-cell lung cancer P-value=0.0232; prostate cancer P-value= 0.0284; pancreatic cancer P-value=0.0355; chronic myeloid leukemia P-value=0.0414; wnt signaling P-value=0.0436; cell cycle P-value=0.0497) (see Supplementary Table 10, online).
Discussion
Our study is the first genome-wide array based analysis of CNAs/cnLOHs that both characterizes and quantifies the proportion of subclones carrying genomic changes before treatment and at subsequent relapse in cancer. The results demonstrate that (i) many CNAs/cnLOHs in CLL are recurrent and therefore non-random events that expand over time owing to Darwinian selective pressure, (ii) selected genes identified in both well-recognized and newly defined MDRs/MORs present plausible candidates for driving disease progression and include those involved in mature B-cell development, DNA damage response (DNA repair, apoptosis and autophagy), tumor progression and familial B-CLL; pathway analysis was consistent with this. The identification of genes in this way will be informative for focused sequencing strategies. (iii) In our cohort there is strong supportive evidence that genomic complexity is associated with poor risk disease. This adds strength to previous studies that indicated a link between genomic complexity or clonal evolution to survival in CLL.30, 36, 38 (iv) OncoSNP is a powerful computational statistical tool that alters fundamentally our ability to investigate clonal architectures and tease apart complex clonal dynamics. Furthermore, our results demonstrate the importance of developing comparable algorithms for application to next generation sequencing data. Although it is likely that in the longer term array technology will be replaced by whole genome sequencing, this study contributes to our understanding of the significance of CNAs/cnLOH in cancer progression. Finally, our results do not support the existence of a single leukemia propagating cell population but instead, shed light on the considerable genomic heterogeneity of cells driving disease progression.
Clinically, the existence of multiple, genetically distinct, subpopulations that escape therapeutic intervention presents formidable challenges for the development of effective treatments for patients with relapsed refractory B-CLL. Therefore, detailed characterization of the molecular basis of the condition and elucidation of the underlying mechanisms of clonal diversity will be essential for future targeted selection of effective therapeutic agents.
References
Lindblad-Toh K, Tanenbaum DM, Daly MJ, Winchester E, Lui WO, Villapakkam A et al. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat Biotechnol 2000; 18: 1001–1005.
Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 2008; 40: 722–729.
Stratton MR, Campbell PJ, Futreal PA . The cancer genome. Nature 2009; 458: 719–724.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008; 322: 1377–1380.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463: 899–905.
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476: 298–303.
Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475: 101–105.
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011; 471: 235–239.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011; 471: 189–195.
Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G et al. A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol 2010; 11: R92.
Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W,, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A,, Eisner MP, Lewis DR,, Chen HS, Feuer EJ, Cronin KA, Edwards BK (eds). SEER Cancer Statistics Review. National Cancer Institute: Bethesda, MD, 1975–2008. http://seer.cancer.gov/csr/1975_2008/, based on November 2010 SEER data submission, posted to the SEER web site, 2011.
CRUK. http://info.cancerresearchuk.org/cancerstats/types/leukaemia/.
Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA 1991; 88: 5413–5417.
Zenz T, Mohr J, Edelmann J, Sarno A, Hoth P, Heuberger M et al. Treatment resistance in chronic lymphocytic leukemia: the role of the p53 pathway. Leuk Lymphoma 2009; 50: 510–513.
Stilgenbauer S, Sander S, Bullinger L, Benner A, Leupolt E, Winkler D et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 2007; 92: 1242–1245.
Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood 2008; 112: 3322–3329.
Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood 2009; 114: 5307–5314.
Stankovic T, Stewart GS, Fegan C, Biggs P, Last J, Byrd PJ et al. Ataxia telangiectasia mutated-deficient B-cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood 2002; 99: 300–309.
Hallek M . First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL study group. Blood (ASH Ann Meet Abstr) 2009; 114: 535.
Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343: 1910–1916.
Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005; 353: 1793–1801.
Bandi N, Zbinden S, Gugger M, Arnold M, Kocher V, Hasan L et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res 2009; 69: 5553–5559.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102: 13944–13949.
Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol 2007; 27: 2240–2252.
Raveche ES, Salerno E, Scaglione BJ, Manohar V, Abbasi F, Lin YC et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007; 109: 5079–5086.
Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 2008; 14: 1271–1277.
Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010; 17: 28–40.
Ouillette P, Erba H, Kujawski L, Kaminski M, Shedden K, Malek SN . Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res 2008; 68: 1012–1021.
Parker H, Rose-Zerilli MJ, Parker A, Chaplin T, Wade R, Gardiner A et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia 2011; 25: 489–497.
Ouillette P, Collins R, Shakhan S, Li J, Li C, Shedden K et al. The prognostic significance of various 13q14 deletions in chronic lymphocytic leukemia. Clin Cancer Res 2011; 17: 6778–6790.
Sellick GS, Wade R, Richards S, Oscier DG, Catovsky D, Houlston RS . Scan of 977 nonsynonymous SNPs in CLL4 trial patients for the identification of genetic variants influencing prognosis. Blood 2008; 111: 1625–1633.
Pfeifer D, Pantic M, Skatulla I, Rawluk J, Kreutz C, Martens UM et al. Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays. Blood 2007; 109: 1202–1210.
Gunn SR, Mohammed MS, Gorre ME, Cotter PD, Kim J, Bahler DW et al. Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia. J Mol Diagn 2008; 10: 442–451.
Kujawski L, Ouillette P, Erba H, Saddler C, Jakubowiak A, Kaminski M et al. Genomic complexity identifies patients with aggressive chronic lymphocytic leukemia. Blood 2008; 112: 1993–2003.
Kay NE, Eckel-Passow JE, Braggio E, Vanwier S, Shanafelt TD, Van Dyke DL et al. Progressive but previously untreated CLL patients with greater array CGH complexity exhibit a less durable response to chemoimmunotherapy. Cancer Genet Cytogenet 2010; 203: 161–168.
Gunnarsson R, Isaksson A, Mansouri M, Goransson H, Jansson M, Cahill N et al. Large but not small copy-number alterations correlate to high-risk genomic aberrations and survival in chronic lymphocytic leukemia: a high-resolution genomic screening of newly diagnosed patients. Leukemia 2010; 24: 211–215.
Gunnarsson R, Mansouri L, Isaksson A, Goransson H, Cahill N, Jansson M et al. Array-based genomic screening at diagnosis and follow-up in chronic lymphocytic leukemia. Haematologica 2011; 96: 1161–1169.
Ouillette P, Collins R, Shakhan S, Li J, Peres E, Kujawski L et al. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood 2011; 118: 3051–3061.
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science 2008; 319: 336–339.
Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M et al. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 2010; 115: 3553–3558.
Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011; 469: 362–367.
Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2011; 469: 356–361.
Sun W, Wright FA, Tang Z, Nordgard SH, Van Loo P, Yu T et al. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res 2009; 37: 5365–5377.
Greenman CD, Bignell G, Butler A, Edkins S, Hinton J, Beare D et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics 2010; 11: 164–175.
Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA 2010; 107: 16910–16915.
Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J et al. Inferring tumor progression from genomic heterogeneity. Genome Res 2010; 20: 68–80.
Letouze E, Allory Y, Bollet MA, Radvanyi F, Guyon F . Analysis of the copy number profiles of several tumor samples from the same patient reveals the successive steps in tumorigenesis. Genome Biol 2010; 11: R76.
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y et al. Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Acknowledgements
This work was supported by the NIHR Biomedical Research Centre, Oxford with funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. In addition, the publication presents independent research commissioned by the Health Innovation Challenge Fund (HICF-1009-026), a parallel funding partnership between the Wellcome Trust and the Department of Health. The views expressed in this publication are those of the authors and not necessarily those of the Wellcome Trust or the Department of Health. This study makes use of data generated by the Wellcome Trust Case–Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. The project was also supported by the Wellcome Trust (090532/Z/09/Z), (076113), (085475) and by Illumina, Inc. CY is funded by a UK Medical Research Council Specialist Training Fellowship in Biomedical Informatics (Ref No. G0701810). We would also like to acknowledge Dr David Bentley (Illumina Inc) and Prof Ian Tomlinson (WTCHG) for their role in supporting this work.
Author contributions
SK designed and performed the research, analyzed the data and wrote the manuscript; RC, ATT, HMD, AB and ESA helped design the research and performed experiments; ARP and DGO provided samples; CY, CCH, JB, CC and SD analyzed data and contributed to the manuscript, JT was responsible for project management and co-ordination and helped write the manuscript, AS provided samples, designed the research, analyzed the data, and wrote and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
We would like to declare that OncoSNP is freely available for use in academic or non-profit research activities upon request. Isis Innovation Ltd may request a fee for the use of the software in commercial or other profit generating activities. It is the policy of Isis Innovation Ltd that a share of the revenue generated from such licensing arrangements would be distributed via the University of Oxford to CY and CCH.
Additional information
Supplementary Information accompanies the paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Knight, S., Yau, C., Clifford, R. et al. Quantification of subclonal distributions of recurrent genomic aberrations in paired pre-treatment and relapse samples from patients with B-cell chronic lymphocytic leukemia. Leukemia 26, 1564–1575 (2012). https://doi.org/10.1038/leu.2012.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2012.13
Keywords
This article is cited by
-
Bootstrap Testing of Central Tendency Nullity Over Paired Fuzzy Samples
International Journal of Fuzzy Systems (2021)
-
Multi-platform profiling characterizes molecular subgroups and resistance networks in chronic lymphocytic leukemia
Nature Communications (2021)
-
Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome
Leukemia (2017)
-
Using Genomic Information to Guide Ibrutinib Treatment Decisions in Chronic Lymphocytic Leukaemia: A Cost-Effectiveness Analysis
PharmacoEconomics (2017)
-
Longitudinal copy number, whole exome and targeted deep sequencing of 'good risk' IGHV-mutated CLL patients with progressive disease
Leukemia (2016)
