
Abstract
PGE2 regulates glomerular hemodynamics, renin secretion, and tubular transport. This study examined the contribution of PGE2 EP1 receptors to sodium and water homeostasis. Male EP1−/− mice were bred with hypertensive TTRhRen mice (Htn) to evaluate blood pressure and kidney function at 8 weeks of age in four groups: wildtype (WT), EP1−/−, Htn, HtnEP1−/−. Blood pressure and water balance were unaffected by EP1 deletion. COX1 and mPGE2 synthase were increased and COX2 was decreased in mice lacking EP1, with increases in EP3 and reductions in EP2 and EP4 mRNA throughout the nephron. Microdissected proximal tubule sglt1, NHE3, and AQP1 were increased in HtnEP1−/−, but sglt2 was increased in EP1−/− mice. Thick ascending limb NKCC2 was reduced in the cortex but increased in the medulla. Inner medullary collecting duct (IMCD) AQP1 and ENaC were increased, but AVP V2 receptors and urea transporter-1 were reduced in all mice compared to WT. In WT and Htn mice, PGE2 inhibited AVP-water transport and increased calcium in the IMCD, and inhibited sodium transport in cortical collecting ducts, but not in EP1−/− or HtnEP1−/− mice. Amiloride (ENaC) and hydrochlorothiazide (pendrin inhibitor) equally attenuated the effect of PGE2 on sodium transport. Taken together, the data suggest that EP1 regulates renal aquaporins and sodium transporters, attenuates AVP-water transport and inhibits sodium transport in the mouse collecting duct, which is mediated by both ENaC and pendrin-dependent pathways.
Similar content being viewed by others
Main
Hypertension is one of the major health issues worldwide, and a cause and consequence of kidney disease. The renin-angiotensin system (RAS) remains the primary therapeutic target in patients with kidney disease.1 Prostaglandin E2 (PGE2) has an important role in RAS activation by stimulating renin secretion, and contributes to blood pressure (BP) regulation by direct pressor effects or by maintaining salt and water balance.2, 3 Inhibition of the PGE2 synthesis enzymes, COX (cyclooxygenases) and mPGES1 (microsomal PGE2 synthase), has hypertensive consequences,4, 5 yet little is known about the involvement of PGE2 in angiotensin-II-dependent hypertension.
PGE2 is the major renal product of COX1 and COX2 acting through a balance of PGE2 receptor (EP)-mediated responses: EP1–4. The contribution of PGE2 to systemic hypertension and kidney disease remains uncertain. We recently reviewed the role of PGE2 in hypertension and diabetes.6 Most of sodium and H2O reabsorption occurs in the proximal tubule (PT) via the sodium-hydrogen exchanger and aquaporin 1. However, the collecting duct (CD) has an important role in the fine regulation of sodium and H2O homeostasis, and is a major site of renal PGE2 action and EP1 expression. PGE2/EP1 increases intracellular Ca2+ through the activation of phospholipase C, to inhibit CD sodium reabsorption.7, 8 Genetic disruption of EP1 does not significantly impair sodium excretion in the healthy mouse; however, EP1−/− display elevated renin and aldosterone consistent with sustained activation of RAS,9 and an impaired pressor response to angiotensin-II.10
In addition to angiotensin-II, arginine vasopressin (AVP) is a key regulator of distal nephron sodium and water transport, and is elevated in hypertension and diabetes.11 Water homeostasis and urine concentration are highly dependent on the CD response to AVP, which stimulates aquaporin-2 mediated water reabsorption via V2 receptors. Though PGE2 can stimulate CD water reabsorption in the absence of AVP via EP4, PGE2 attenuates AVP-mediated transport through EP3 in the rat and rabbit.12, 13 Inner medullary CD display blunted AVP-water flux but enhanced urea permeability in response to PGE2.14, 15
We have recently demonstrated a role for PGE2/EP1 in PT water transport,16 and confirmed that a disturbance in EP3 function only partially contributes to defective urine-concentrating ability in diabetic mice,17 highlighting a possible involvement of EP1 in kidney concentrating function. Accumulating evidence suggests that a sustained activation of the local RAS within the CD has a key role in the development of angiotensin-II-dependent hypertension.18, 19, 20 Accordingly, the purpose of the work was to study the transport properties of the CD in hypertensive mice, and determine the role of renal PGE2/EP1 in angiotensin-II-dependent hypertension.
First, we examined the expression of the PGE2/EP system in microdissected nephron segments from TTRhRen hypertensive mice (a model of angiotensin-II-dependent hypertension), EP1−/−, and hypertensive mice lacking EP1. We also measured the expression of major renal sodium transporters and water channels, as well as AVP receptors and urea transporters in the kidney in these mice. Finally, we explored the role of EP1 in CD salt and water transport. Our work demonstrates that EP1 has a major role in inhibiting AVP-water transport in the mouse IMCD and CCD, in addition to decreasing transepithelial voltage (reduced sodium transport) in isolated perfused mouse CCD, but does not impact BP in young TTRhRen mice.
Materials and methods
Animal procedures were approved by the University of Ottawa Animal Care Service in accordance with the Canadian Council on Animal Care (CCAC) guidelines. FVB/n mice were housed on a 12-h light-dark cycle, with free access to standard chow and water. Male mice were studied at 8 weeks of age. We have previously characterized global EP1−/− and hyperreninemic hypertensive TTRhRen (Htn) mice following diabetes induction.21, 22 In this study, EP1−/− were bred with Htn. Four groups were studied: wildtype (WT), EP1−/−, Htn, and HtnEP1−/−, n=12–15 mice per group.
Physiological Parameters, BP, Metabolic Cages, and Urine Analysis
Systolic BP was measured at the same time daily by tail-cuff plethysmography (BP 2000, Visitech systems, Apex, NC, USA). Following a five-day training period, the average of five consecutive days was calculated as described.23 Mice were placed in metabolic cages with free access to water and food. Twenty-four hour urine output and water intake were recorded following a 24-h adaptation period. Urine osmolality was determined by freezing point depression (Advanced Model 3MO Plus Osmometer, Advanced Instruments, Norwood, MA, USA). AVP excretion rates were measured using Arg8-Vasopressin ELISA (Enzo Life Sciences, Farmingdale, NY, USA) and corrected for 24-h urine output.
Glomerular Filtration Rate
Glomerular filtration rate (GFR) was estimated in conscious mice by fluorescein isothiocyanate-labeled (FITC)-inulin clearance (Sigma-Aldrich, Oakville, ON, USA) as described.17, 24 Mice were injected with FITC-inulin (3.74 μl/g BW; Sigma-Aldrich, St Louis, MO, USA) via tail vein. Blood was collected at timed intervals from the saphenous vein into heparinized capillary tubes and centrifuged. Samples were buffered in HEPES (500 mM, pH 7.4) and fluorescence was measured (excitation 488 nm/emission 538 nm). GFR was calculated using a two-compartment clearance model.24
Quantitative PCR
Kidneys were dissected to separate cortex and medulla as described.23 Snap frozen tissue was homogenized using the TP-103 Amalgamator COE Capmixer (GC America, Alsip, IL, USA). RNA was extracted with TRIzol and treated with DNAse I (Invitrogen). mRNA was measured by quantitative PCR (qPCR) with the ABI Prism 7000 system using specific primers as described,25 and the SYBR Advantage qPCR Premix (Clontech Laboratories, Mountain View, CA, USA) according to the manufacturer’s instructions. Expression was normalized to 18 S RNA and the 2(-DeltaDeltaC(T)) method was used for analysis.26 For microdissected PT, thick ascending limb (TAL), CCD and IMCD, RNA from 6 to 8 tubules was extracted using Qiagen RNEasy minikit, and reverse transcribed using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). Table 1 lists the primers used for qPCR.
In Vitro Microperfusion
Mice were killed at 8 weeks of age. The CCD and IMCD were microdissected for in vitro microperfusion and the measurement of net fluid reabsorption (Jv) as described.16, 17 Briefly, the bath solution was exchanged at 0.5 ml/min and maintained at 37 °C. For the CCD, the perfusate was hypotonic at 180 mOsm vs 290 mOsm for the bath; and the perfusate for the IMCD was isotonic at 290 mOsm vs 440 mOsm for the bath. The perfusion rate of 12–20 nl/min was maintained by adjusting the hydrostatic pressure. 3H-inulin (75 μCi/ml) was used as a volume marker. During control periods, two collections were made for the calculation of basal Jv following 30 min equilibration. Tubules with a negative basal Jv were discarded. AVP: 10-8 (CCD) or 10-12 (IMCD) mmol/l (Sigma-Aldrich, St Louis, MO, USA) was added to the bath and four collections were made. These concentrations were selected after testing the responsiveness of tubules taken from wildtype mice, with a range of AVP concentrations from 10-5 mmol/l to 10-12 mmol/l. Either PGE2 or the EP1/3 agonist sulprostone (SLP) was then added to the bath (10-7 mmol/l, Sigma-Aldrich, St Louis, MO, USA) with five additional collections. The mean Jv was calculated as the difference between perfusion rate Vo and collection rate VL, in nl/min, normalized to tubule length (L, in mm): Jv=(Vo−VL)/L, where Vo=VL(CL/Co), and where CL and Co are perfusate and collected fluid concentrations in cpm/nl, respectively. To further confirm the involvement of EP1, IMCDs were taken from C57Bl/6 EP3−/− mice and microperfused to determine AVP-Jv in response to sulprostone.
Intracellular Calcium
Microdissected IMCDs were loaded with 1 μM Fura-2AM (Molecular probes) for 30 min at 30 °C. Tubules were perfused with Ca2+ and Mg2+-free hypertonic perfusate containing 50 μM EGTA. Fura-2AM was washed away using bath solution and the temperature was increased to 37 °C. The bath exchange rate was maintained at 1.5 ml/min. Intracellular Fura-2AM fluorescence intensity at 520 nm was measured by photon counting using the Deltascan System (Photon Technology International, New Brunswick, NJ, USA), with continuous rapidly alternating excitation wavelengths of 340 and 380 nm; output was coupled to an inverted microscope through a 400 nm dichroic mirror as described.14 The ratio of emission intensity at 340 and 380 nm (340/380 ratio) was continuously recorded for each experiment. Following a 5 min baseline recording, the tubule was stimulated with 1 μM PGE2 for 3 min, then washed with bath solution, and stimulated for 3 min with 1 μM AVP, followed by bath solution.
Transepithelial Voltage
Microdissected CCDs were cannulated as described above. Two probes were used to measure the transepithelial voltage across the tubule, from lumen to bath, using an electrometer set at continuous readings for 20 min (WPI, KS-700). The response to 1 μM PGE2 was studied. Also, tubules were pre-incubated with 10 μM amiloride or hydrochlorothiazide to inhibit epithelial sodium channels (principal cell ENaC) and intercalated cell pendrin-dependent transport (via inhibition of carbonic anhydrase), respectively, prior to PGE2 stimulations.19 The delta transepithelial voltage was calculated by subtracting PGE2 stimulations from baseline in the presence or absence of each inhibitor. An inhibition of sodium transport by PGE2 is observed as depolarization of the transepithelial voltage.
Statistics
Graphpad Prism (San Diego, CA, USA) was used for analysis. Values are expressed as means±s.e.m. Statistical analysis was done using one-way ANOVA followed by Tukey’s post-test, and by unpaired t-test for comparisons between two groups. In addition, a one sample t-test with a hypothetical value of 1 was performed for qPCR.
Results
Physiological Parameters, BP, and GFR
To study the effect of chronic hypertension and EP1 receptor deletion on renal function, we measured glomerular filtration rate, kidney weights, volume status, and urine properties. Body and kidney weights were significantly increased in EP1−/− and HtnEP1−/− compared to WT (Table 2). However, kidney weights remained significantly higher only for EP1−/− when normalized to tibia length. Urine volume was increased in all groups compared to WT, up to twofold in HtnEP1−/−, but was not statistically significant. Similarly, water intake was increased in all groups compared to WT, but only reached significance in HtnEP1−/− at 5.1 ml/day compared to 3.1 ml/day in WT.
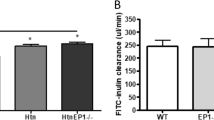
GFR and BP were unaffected by EP1 deletion. In Figure 1a, systolic BP was significantly higher in Htn and HtnEP1−/− (150 mm Hg) compared to WT (125 mm Hg), but no difference was observed between Htn and HtnEP1−/−. There was no significant difference in FITC-inulin clearance across the four groups, calculated at 200 μl/min (Figure 1b). Urine osmolality was unchanged in normotensive EP1−/−, but was significantly reduced in hypertensive mice from 2500 mOsm/KgH2O in WT to 1681 in HtnEP1−/− (Figure 1c). No difference was noted between Htn and HtnEP1−/− osmolalities. Urinary AVP excretion rates were increased twofold in both hypertensive groups, but were significantly different only in Htn, and unaltered by EP1 deletion (Figure 1d). The AVP levels were not significantly different between EP1−/− and HtnEP1−/−, though there is a twofold difference between the two groups.

Systolic blood pressure BP (a), FITC-inulin clearance (b), urine osmolality (c), and urinary vasopressin (AVP) excretion rates (d) were assessed in four mouse groups: wildtype (WT), EP1 receptor null mice (EP1−/−), hypertensive TTRhRen mice (Htn), and hypertensive EP1 null mice (HtnEP1−/−). Data are presented as mean±s.e.m., n=7–9. * indicates significance vs WT. P<0.05, and bar indicates significance between two groups.
Renal Cyclooxygenase, PGE2 Synthase, and EP Receptors
To determine whether the PGE2 system is altered in hypertensive kidneys or in response to EP1 receptor deletion, and examine how and where these changes occur in the kidney, we characterized the regional and segmental mRNA expression profile of the PGE2 pathway, including the synthetic enzymes: COX1 and COX2 and PGE2 synthases (microsomal mPGES1 and cytosolic cPGES), as well as the four PGE2 EP receptors (EP1–4).
As shown in Table 3, cortical COX2 mRNA was significantly reduced by over 50% in all groups compared to WT. Medullary COX2 mRNA levels were different in each group: reduced by 45% in EP1−/−, unchanged in Htn, and increased threefold in HtnEP1−/−; but not statistically significant. Cortical mPGES1 mRNA was significantly increased 1.5- and 1.7-fold in Htn and HtnEP1−/−, respectively. The increase in cortical mPGES1 mRNA could be a compensatory increase (because of COX2 reduction) that is not EP1 dependent. Medullary mPGES1 mRNA was also increased in all groups compared to WT, though a great deal of variability was observed and not statistically significant. cPGES mRNA was unchanged.
PT COX1 was significantly increased threefold in HtnEP1−/− compared to the three other groups, and unchanged in other groups compared to WT. COX1 was also increased in all three groups compared to WT in the TAL, CCD and medulla; but was significant only in the following groups: 2.5-fold in the cortical TAL of EP1−/− and HtnEP1−/−; fourfold in the CCD of HtnEP1−/−, and twofold in the medulla of Htn and HtnEP1−/−, with no significant difference between Htn and HtnEP1−/− COX1 mRNA levels.
PT EP1 mRNA was increased in all three groups compared to WT reaching 20-fold in HtnEP1−/−, but was not statistically significant in EP1−/−. CCD EP1 was significantly increased 2.5-fold in HtnEP1−/−, but EP1 was unchanged in the TAL and reduced up to 30% in the IMCD of mice lacking EP1. The IMCD reduction was only significant in EP1−/− and HtnEP1−/−. As described by Guan et al.,10 the EP1−/− mouse was generated using a ‘hit-and-run’ strategy to disrupt the gene encoding EP1 but spare protein kinase N (PKN), which is encoded at the EP1 locus on the antiparallel DNA strand. Using this strategy, a premature in-frame stop codon and EcoRI site were introduced into the mutant EP1 mRNA, which can be digested and revealed by PCR. For consistency, we performed qPCR on all the mice in our study, and the mRNA increases in the EP1−/− mice are likely due to feedback responses compensating for the absence of functional EP1 protein. EP3 was increased in PT, CCD and IMCD of mice lacking EP1, possibly compensating for the lack of EP1. PT EP3 was increased twofold in Htn and EP1−/−, and fivefold in the HtnEP1−/− compared to WT, though not significant. In the CCD and IMCD, EP3 was significantly increased in EP1−/− and HtnEP1−/− up to fourfold compared to WT, but unchanged in Htn. Like EP1, EP3 was unchanged in the TAL. However, EP4 was significantly reduced by over 60% in the TAL and CCD in all three groups compared to WT. In contrast, PT and IMCD EP4 was significantly increased threefold in HtnEP1−/−, and 2.5-fold in EP1−/− IMCD compared to WT, again possibly compensating for the lack of EP1. Medullary EP1 was significantly increased by 10-fold in all groups compared to WT, whereas EP2 was significantly reduced by 50% in Htn and HtnEP1−/−. In comparison, in the cortex segments, EP1 was increased in the PT of all three groups compared to WT, and only in the CCD of HtnEP1−/− mice. EP4 was also significantly reduced by 45% in all groups compared to WT, similar to reductions observed in the TAL and CCD in the cortex. Medullary EP3 was unchanged, whereas it was significantly increased in the PT and CCD of the cortex. EP2 was not detectable in the PT, TAL or CCD (not shown). Of interest, the pattern of alterations in expression of EP receptor mRNA in the whole medulla sample were often opposite those observed in the microdissected terminal IMCD, highlighting the possibility that differences in EP receptor regulation are occurring in different cells/segments of the nephron, according to the underlying functions. But more work is needed to uncover these specific roles of the PGE2/EP system in the different parts of the nephron. Another possibility is that other prostaglandin systems are modified in response to changes in COX or EP receptors in hypertensive kidneys or in mice lacking EP1, and this opens up a plethora of avenues to explore.
Renal Sodium Transporters and Water Channels
To study the effect of chronic hypertension and EP1 receptor deletion on renal sodium and water transporters, and determine which segment is dependent on EP1 receptor expression for sodium and water transport in hypertensive kidneys, we examined the mRNA expression of major sodium transporters and apical aquaporins in various kidney preparations. As shown in Table 4, sodium-hydrogen exchanger (NHE)-3 was unchanged in EP1−/− and Htn PT, but significantly increased fourfold in HtnEP1−/−, compared to WT. Similarly, sodium glucose co-transporter (sglt)-1 was significantly increased by 2.7-fold in HtnEP1−/− PT compared to WT. Although sglt2 was unchanged in hypertensive mice; it was significantly increased twofold in EP1−/− PT compared to WT. PT aquaporin (AQP)-1 was also increased to 2.7-fold in all groups compared to WT, but not significant in EP1−/−.
Sodium-potassium-2 chloride co-transporter (NKCC2) was significantly reduced by 40% in EP1−/− cortex, and increased in EP1−/− medulla by 1.7-fold, though not significant compared to WT. NKCC2 was unchanged in other mice. Epithelial sodium channel (αENaC) was reduced by 20% in EP1−/− cortex, though not significant, but significantly increased in Htn cortex compared to WT. Medulla αENaC was unchanged.
As shown, V2R is unchanged in the cortex (1.5-fold) but significantly increased by 2.3-fold in the EP1−/− medulla (twofold) compared to WT, but not in hypertensive mice. However, AQP2 is significantly increased 1.5-fold in the Htn cortex compared to WT, but increased twofold in the medulla of EP1−/− compared to WT, though not significant. The cortical induction of AQP2 in hypertensive mice is significantly attenuated by EP1 deletion.
In microdissected terminal IMCD AQP2 was reduced by 45% in Htn, though not statistically significant, and unchanged in mice lacking EP1. However, V2R and urea transporter UTA1 were significantly reduced in all groups compared to WT up to 50 and 80% respectively. In contrast, αENaC was significantly augmented by 2–4-fold in all groups compared to WT. The cortico-medullary osmotic gradient partly depends on AQP1 in the medullary thin descending limbs and vasa recta.27 As shown, inner medullary AQP1 was increased up to 2-fold in all groups compared to WT, but was not significant in Htn. Altogether this suggests that not only is EP1 having key role in inhibiting IMCD water and sodium transporter mRNA expression (which is consistent with its role in inhibiting sodium and water transport in the CD), but it also regulates basal NKCC2 expression and aquaporin 1 in the loop of Henle to affect medullary concentration needed for CD water reabsorption.
Water Flux, Intracellular Calcium, and Transepithelial Voltage
To study the transport properties of the collecting duct in response to chronic hypertension and EP1 receptor deletion, we measured water reabsorption in the IMCD and CCD, as well as intracellular calcium and transepithelial voltage in the CCD, in response to PGE2 stimulation. PGE2 inhibits AVP-fluid reabsorption in microdissected terminal IMCD and CCD. A representative tracing of Jv over time is shown for one IMCD from each group (Figure 2a). AVP increased net fluid reabsorption (Jv) to 3–5 nl/mm/min in IMCD from all groups, but the response was significantly greater in EP1−/− compared to WT (Figure 2b). PGE2 significantly attenuated this response in WT and Htn, but not in mice lacking EP1. Similarly, the EP1/3 agonist sulprostone (SLP) significantly reduced AVP-Jv in WT IMCD, but not EP1−/− (Figure 2c). In contrast, SLP attenuated AVP-Jv in EP3−/− IMCD (Figure 2d). In comparison, PGE2 only partially attenuated CCD AVP-Jv in WT and mice lacking EP1 (Figure 2f), but was not significant.

Net fluid reabsorption (Jv) was analyzed in four mouse groups: wildtype (WT), EP1 receptor null mice (EP1−/−), hypertensive TTRhRen mice (Htn), and hypertensive EP1 null mice (HtnEP1−/−). The terminal inner medullary collecting ducts (a–d) and CCD: cortical collecting ducts (e) were microdissected for in vitro microperfusion and determination of Jv (nl/mm/min) upon stimulations with vasopressin (AVP: white bars): 10−12 M for terminal inner medullary collecting ducts (a–d) and 10−7 M for CCD: cortical collecting ducts (e). Tubules were also stimulated with AVP followed by 10−7M PGE2 (AVP+PGE2: black bars) or 10−7 M sulprostone (AVP+SLP: black bars). A representative time course in minutes (min) of Jv is shown (a) as well as bar graphs of mean±s.e.m. (b–e), n=5. * indicates significance vs AVP alone, P<0.05. Bar indicates significance between two groups.
PGE2 increased intracellular calcium (increased 340/380 ratio) in WT FVB (Figure 3a) and C57Bl/6 (Figure 3b) mice, but not in mice lacking EP1 (Figure 3c). In EP3−/−, the response was like WT (Figure 3d). In comparison, AVP increased calcium in all mouse groups (Figures 10a-f). The delta 340/380 ratio diminished significantly from 0.420±0.040 in WT FVB mice stimulated with PGE2 to 0.050±0.004 in EP1−/− (Figure 3e).

Intracellular calcium was measured in isolated perfused inner medullary collecting ducts following loading with Fura-2AM, in four mouse groups: WT FVB (a), WT C57Bl/6 (b), EP1−/− (c), and EP3−/− (d). For each experiment, following a baseline recording, the 340/380 nm excitation ratios were recorded in response to 1 μM PGE2 stimulations (indicated with bar), followed by a washout period, then stimulations with 1 μM AVP (indicated with bar). A representative tracing of the absolute 340/380 nm ratio for each is shown (a–d). The 340/380 ratios for each stimulation were subtracted from baseline and the average of three experiments was calculated for FVB and EP1−/− mice (e) and C57Bl/6 and EP3−/− mice (f). Data are presented as mean±s.e.m. of the delta ratio (stimulation-baseline), n=3–4, *P<0.05.
Finally, PGE2 depolarized the CCD lumen to bath voltage, from −35 to −25 mV in WT, a representative tracing from each group is shown (Figures 4a and b). A similar response was observed in Htn, but was significantly abolished in mice lacking EP1 (Figure 4b). The PGE2-depolarization was significantly diminished by 54% with the pendrin inhibitor hydrochlorothiazide and 46% by the ENaC inhibitor amiloride, though not statistically significant for the latter (Figure 4c), suggesting that PGE2 inhibits sodium transport in the CCD via both pathways. It is noteworthy that hydrochlorothiazide indirectly implicates pendrin in EP1-mediated sodium inhibition, supported by the recent findings that ENaC and pendrin are equally important in mediating CCD sodium transport,19 and more work is needed to ascertain its direct involvement.

Transepithelial voltage (a–c) in response to 10−7 PGE2 stimulations was determined in cortical collecting ducts from four mouse groups: wildtype (WT), EP1 receptor null mice (EP1−/−), hypertensive TTRhRen mice (Htn), and hypertensive EP1 null mice (HtnEP1−/−). A representative time course in minutes (min) is shown (a), for baseline followed by PGE2 stimulations (indicated with black line). A bar graph of mean±s.e.m. of transepithelial voltage (−mV), n=5 is shown (b). The effect of two diuretics on PGE2-stimulated transepithelial voltage presented as delta (Δ) transepithelial voltage is shown, amiloride (P+amiloride) and hydrochlorothiazide (P+HCZ), n=5. * indicates significance vs WT with P<0.05, and bar indicates significance between two groups.
Discussion
This study demonstrates a major role for EP1 in mouse CD sodium and water transport, and is the first to examine the PGE2/EP system in hypertensive mouse kidneys. We have novel data showing that PGE2/EP1 attenuates AVP-H2O reabsorption in mouse IMCD and PGE2/EP1 inhibits sodium transport by both ENaC and pendrin-dependent pathways.
Generally, COX1 and mPGES1 were elevated in all nephron segments of EP1−/− except COX1 in the PT. Altogether our data suggest that COX1 mRNA expression is EP1 dependent in the TAL and CCD, or may be even that PGE2/EP1 acts as a tonic inhibitor of COX1 in these segments, but not in the PT. Cortical COX2 was reduced perhaps by negative feedback,28 considering the sustained activation of RAS in Htn and the role of macula densa COX2 in activating RAS.29 COX2 was also reduced in EP1−/−, suggesting EP1 may be involved in its regulation. Whether the effect directly involves EP1 or is due to RAS activation in these mice is not clear at this time. Consistent with involvement of RAS is the fact that the reduction of COX2 was also seen in Htn mice, and angiotensin-II is known to regulate COX2. This is interesting considering that we showed that COX1 and COX2 were unchanged in EP3−/−.17 Though not much is known about the signals that trigger COX2 downregulation, Haddad et al.30 demonstrated that EP1 may promote COX2 ubiquitination and subsequent degradation. In the medulla, the data suggest that both COX1 and COX2 are downregulated by PGE2/EP1, but not as simple to interpret at this time, considering that Htn mice also display similar changes as HtnEP1−/− with respect to COX1 mRNA expression, yet EP1 mRNA levels are significantly elevated in Htn medulla. Furthermore COX2 is decreased in EP1−/− medulla, but increased in HtnEP1−/− medulla. Clearly, there is a complex interaction between PGE2/EP systems and RAS, and perhaps other hormonal systems like AVP, that determine the overall effect on COX expression, PGE2 synthesis, and EP receptor signaling. More work is needed to fully appreciate this multifaceted interplay. Consistent with previous reports that cPGES does not contribute much to renal PGE2 synthesis,31 cPGES was unchanged in our study.
The EP profile was also altered. Overall, EP3 was increased in mice lacking EP1, but cortical EP4 and medullary EP2 and EP4 were reduced. In the PT both EP3 and EP4 were elevated, most likely compensating for a lack of EP1. In the TAL, only EP4 was reduced; not surprising given that like COX2, EP4 mediates renin stimulation,29, 32 with EP4 reduction due to feedback inhibition. Though CD EP3 was reduced, there were notable differences in EP4 expression: reduced in CCD but increased in IMCD. The reason for these differences is not clear, but disruption of mouse CD EP4 resulted in altered AQP2 membrane distribution and defective urinary concentrating function, identifying a central role for EP4 in AVP-independent water homeostasis.33 Of importance, renal EP profiles were unchanged in EP3−/−.17
We recently revealed a central role of EP3 in urinary concentrating mechanisms using streptozotocin-diabetic EP3−/−.17 We also showed that the CD is subjected to injury when exposed to increased filtrate flow associated with polyuria, and revealed a partial role for EP3 in this injury.17 Though we did observe polyuria in our hypertensive mice, there were no signs of glomerular or tubular injury (not shown), and overall tubular function seems intact with similar transport responses. The EP3 study was conducted following 12 weeks of diabetes induction in 20-week-old mice, thus older Htn mice may exhibit more pronounced signs of CD injury associated with prolonged exposure to hypertension and increased luminal flow, and therefore defective tubular transport properties may be observed. Ongoing studies are being conducted to examine the role of EP1 in the older hypertensive-diabetic kidney. Of note, we previously reported a key role for EP1 receptors in hypothalamic release of AVP in the setting of acute water deprivation in mice, revealing a urine-concentrating defect in EP1−/− mice that were water deprived for 24 h, with significant reductions in urine AVP excretion.34 In contrast, our study indicates elevated levels of AVP in Htn mice that were unchanged by EP1 deletion.
In addition, aquaporins and sodium transporter expression were altered throughout the nephron. NHE3, sglt1, sglt2, and AQP1 were variably increased in microdissected PT, strongly suggesting that EP1 may be important in fine-tuning PT sodium reabsorption in hypertension via regulation of these transporters. To the best of our knowledge, the role of EP1 in the PT has not been studied, but we previously reported that PGE2-water transport in the isolated perfused mouse PT was dependent on EP1 under physiological conditions16 and further studies are needed to fully explore its role. In comparison, we recently showed that neither AQP1 nor AQP2 were altered in the EP3−/− kidney.17
PGE2 inhibits medullary TAL NKCC2,35 and in our study cortical NKCC2 was reduced but increased in the medulla in mice lacking EP1. This seems consistent with the hypothesis that PGE2/EP1 has two different roles in the cortex and medulla, i.e. macula densa sensing mechanism in the cortex, and interstitial sodium concentration in the medulla. Clearly, EP1 may be implicated in both these functions which warrants further investigation. In a model of nephrogenic diabetes insipidus, COX2 inhibition ameliorated polyuria by increasing NKCC2 and AQP2,36 further supporting the idea that PGE2 acts as a tonic inhibitor of NKCC2,34 and PGE2 helps maintain water balance by actions on most, if not all, renal sodium transporters. Interestingly, EP3 downregulates medullary TAL COX2 in hypertonic states, providing a means to limit TAL sodium transport in response to high tonicity.37 More work is needed to fully appreciate the respective roles of EP1 and EP3 in TAL sodium transport in both normotensive and hypertensive states.
Angiotensin-II stimulates pituitary AVP when plasma osmolality increases.38 In the context of sustained stimulation of AVP irrespective of plasma osmolality status, as seen in our Htn, the CD responsiveness to AVP may be modified to maintain water and salt balance. Consistent with this notion, urinary AVP was increased, but IMCD V2Rs were diminished. AVP/V2R also increases urea transporter A1 (UTA1) in the IMCD to maintain urine-concentrating function.39 Consistently UTA1 was reduced in microdissected terminal IMCD, including those from normotensive EP1−/−. Though a reduced responsiveness of the CD to AVP may contribute to polyuria, we did not detect any differences in AVP-regulated water transport in the terminal IMCD of hypertensive mice. As such, carrying out similar studies in older TTRhRen mice may reveal a role for EP1 in kidney injury and tubular transport dysfunction.
The role of PGE2 in CD AQP2 expression has been inconsistent. It seems PGE2 does not alter AQP2 levels but affects the AVP-dependent distribution of AQP2. The mechanisms remain unclear, but in the rat PGE2-stimulated AQP2 endocytosis.40 PGE2/EP1 stimulated AQP2 internalization in frog bladder,41 but in the CD, EP3 inhibited AVP-AQP2 membrane shuttling.42 We previously reported no change in V2R, UTA1, or AQP2 in EP3−/− kidneys,17 but more work is needed to decipher the underlying roles of EP1 and EP3 in AVP-dependent or independent CD water transport. Of note, we observed different patterns of V2R, AQP2, and αENaC mRNA expression in the terminal IMCD compared to whole medulla. The medulla preparation contains the outer medullary CD, initial IMCD, and terminal IMCD, and these sub-segments clearly have distinct functionality,43 but their respective roles and responses with respect to electrolyte, water and urea transport are not fully appreciated. In unpublished work, we observed that PGE2 increased Na-K-ATPase protein in terminal IMCD3 cells but not in initial IMCDK2 cells. Experiments were currently performed on the IMCD tip; however, it would be interesting to compare the PGE2 responses of each sub-segment. To the best of our knowledge, this has not been done for any of the major CD hormonal systems, eg, AVP or angiotensin-II.
Our study is the first to describe a functional role of EP1 in mouse CD fluid reabsorption and sodium transport. We showed that EP1 and not EP3 attenuates AVP-fluid reabsorption in the mouse IMCD, whereby PGE2 and sulprostone reverse AVP-fluid reabsorption in only EP1+/+ but not EP1−/−. There is also a notable increase in AVP-fluid reabsorption in normotensive EP1−/−, suggesting EP1 may determine the basal responsiveness of the CD to AVP. Furthermore, we demonstrated that EP1 increases intracellular calcium in mouse IMCD, but EP3 deletion does not affect PGE2-mediated calcium responses. We also confirmed a role for EP1 in CCD sodium transport; but more importantly that PGE2/EP1 equally inhibits ENaC and pendrin-dependent transport. It is important to note that we have indirectly demonstrated pendrin involvement with hydrochlorothiazide, supported by the recent findings that ENaC and pendrin are equally important in mediating CCD sodium transport,19, 44 but more work is needed to ascertain its direct involvement. Though we report a primary role for PGE2/EP1 in the physiological regulation of transporters in the PT and CD, our hypertensive mice did not show altered CD transport function. Perhaps at this early stage, compensatory mechanisms are working to ensure adequate renal handling of sodium and water, and only in prolonged hypertensive states will defective renal transport be a contributing factor.
References
James PA, Oparil S, Carter BL et al. Evidence-based guideline for the management of high blood pressure in adults report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014;311:507–520.
Nasrallah R, Hassouneh R, Hébert RL . Chronic kidney disease: targeting prostaglandin E2 receptors. Am J Physiol Renal Physiol 2014;307:F243–F250.
Swan CE, Breyer RM . Prostaglandin E2 modulation of blood pressure homeostasis: studies in rodent models. Prostaglandins Other Lipid Mediat 2011;96:10–13.
Gurwitz JH, Avorn J, Bohn RL et al. Initiation of antihypertensive treatment during nonsteroidal anti-inflammatory drug therapy. JAMA 1994;272:781–786.
Yang T . Microsomal prostaglandin E synthase-1 and blood pressure regulation. Kidney Int 2007;72:274–278.
Nasrallah R, Hassouneh R, Hébert RL . Prostaglandin E2, kidney disease, and cardiovascular risk: beyond hypertension and diabetes. J Am Soc Nephrol 2016;27:666–676.
Hébert RL, Jacobson HR, Breyer MD . PGE2 inhibits sodium transport in the rabbit CCD by raising intracellular calcium. J Clin Invest 1991;87:1992–1998.
Guan Y, Zhang Y, Breyer RM et al. PGE2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest 1998;102:194–201.
Stock JL, Shinjo K, Burkhardt J et al. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest 2001;107:325–331.
Guan Y, Zhang Y, Wu J et al. Antihypertensive effects of selective prostaglandin E2 receptor subtype 1 targeting. J Clin Invest 2007;117:2496–2505.
Bankir L, Bouby N, Ritz E . Vasopressin: a novel target for the prevention and retardation of kidney disease? Nat Rev Nephrol 2013;9:223–239.
Hébert RL, Jacobson HR, Fredin D et al. Evidence that separate PGE2 receptors modulate water and sodium transport in rabbit cortical collecting duct. Am J Physiol 1993;265:F643–F650.
Olesen ET, Rutzler MR, Moeller HB et al. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc Natl Acad Sci USA 2011;108:12949–12954.
Nadler SP, Zimpelmann JA, Hébert RL . PGE2 inhibits water permeability at a post-cAMP site in rat terminal inner medullary collecting duct. Am J Physiol 1992;262:F229–F235.
Rouch AJ, Kudo LH . Role of PGE2 in alpha2-induced inhibition of AVP- and cAMP-stimulated H2O, Na+, and urea transport in rat IMCD. Am J Physiol Renal Physiol 2000;279:F294–F301.
Nasrallah R, Hassouneh R, Zimpelmann J et al. Prostaglandin E2 increases proximal tubule fluid reabsorption, and modulates cultured proximal tubule cell responses via EP1 and EP4 receptors. Lab Invest 2015;95:1044–1055.
Hassouneh R, Nasrallah R, Zimpelmann J et al. PGE2/EP3 inhibits water reabsorption and contributes to polyuria and kidney injury in streptozotocin diabetic mice. Diabetologia 2016;59:1318–1328.
Chen D, Stegbauer J, Sparks MA et al. Impact of angiotensin type 1 A receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension 2016;67:1291–1297.
Wall SM . The role of pendrin in blood pressure regulation. Am J Physiol Renal Physiol 2016;310:F193–F203.
Prieto MC, Gonzalez AA, Navar LG . Evolving concepts on regulation and function of renin in distal nephron. Pflugers Arch 2013;465:121–132.
Thibodeau JF, Nasrallah R, Hébert RL et al. Reduced albuminuria in diabetic PTGER1-null mice. Am J Pathol 2013;183:1789–1802.
Thibodeau JF, Holterman CE, Burger D et al. A novel mouse model of advanced diabetic kidney disease. PLoS ONE 2014;9:e113459.
Nasrallah R, Robertson SJ, Karsh J et al. Celecoxib modifies glomerular basement membrane, mesangial area and podocyte structure in OVE26 mice, but ibuprofen is more detrimental. Clin Sci 2013;124:685–694.
Qi Z, Whitt I, Mehta A et al. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol 2004;286:F590–F596.
Nasrallah R, Xiong H, Hébert RL . Renal prostaglandin E2 receptor (EP) expression profile is altered in streptozotocin and B6-Ins2Akita type I-diabetic mice. Am J Physiol 2007;292:F278–F284.
Schmittgen TD, Livak KJ . Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101–1108.
Nielsen S, Frokiaer J, Marples D et al. Aquaporins in the kidney: from molecules to medicine. Physiol Rev 2002;82:205–244.
Schnermann J, Briggs JP . Tubular control of renin synthesis and secretion. Pflugers Arch 2013;465:39–51.
Peti-Peterdi J, Harris RC . Macula Densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol 2010;21:1093–1096.
Haddad A, Flint-Ashtamker G, Minzel W et al. Prostaglandin EP1 receptor down regulates expression of cyclooxygenase-2 by facilitating its proteasomal degradation. J Biol Chem 2012;287:17214–17223.
Yang T, Du Y . Distinct roles of central and peripheral prostaglandin E2 and EP subtypes in blood pressure regulation. Am J Hypertens 2012;25:1042–1049.
Pöschke A, Kern N, Maruyama T et al. The PGE2-EP4 receptor is necessary for stimulation of the renin-angiotensin-aldosterone system in response to low dietary salt intake in vivo. Am J Physiol Renal Physiol 2012;303:F1435–F1442.
Gao M, Cao R, Du S et al. Disruption of prostaglandin E2 receptor EP4 impairs urinary concentration via decreasing aquaporin 2 in renal collecting ducts. Proc Natl Acad Sci USA 2015;112:8397–8402.
Kennedy CR, Xiong H, Rahal S et al. Urine concentrating defect in prostaglandin EP1-deficient mice. Am J Physiol Renal Physiol 2007;292:F868–F875.
Kaji DM, Chase HS Jr, Eng JP et al. Prostaglandin E2 inhibits Na-K-2Cl cotransport in medullary thick ascending limb cells. Am J Physiol Cell Physiol 1996;271:C354–C361.
Kim GH, Choi NW, Jung JY et al. Treating lithium-induced nephrogenic diabetes insipidus with a COX-2 inhibitor improves polyuria via upregulation of AQP2 and NKCC2. Am J Physiol Renal Physiol 2008;294:F702–F709.
Hao S, Hernandez A, Quiroz-Munoz M et al. PGE2 EP3 receptor downregulates COX-2 expression in the medullary thick ascending limb induced by hypertonic NaCl. Am J Physiol Renal Physiol 2014;307:F736–F746.
Li XC, Shao Y, Zhuo JL . AT1a receptor knockout in mice impairs urine concentration by reducing basal vasopressin levels and its receptor signaling proteins in the inner medulla. Kidney Int 2009;76:169–177.
Fenton RA . Essential role of vasopressin-regulated urea transport processes in the mammalian kidney. Pflugers Arch 2009;458:169–177.
Zelenina M, Christensen BM, Palmér J et al. Prostaglandin E2 interaction with AVP: effects on AQP2 phosphorylation and distribution. Am J Physiol Renal Physiol 2000;278:F388–F394.
Bachteeva V, Fock E, Lavrova E et al. Prostaglandin E2 inhibits vasotocin-induced osmotic water permeability in the frog urinary bladder by EP1-receptor-mediated activation of NO/cGMP pathway. Am J Physiol Regul Integr Comp Physiol 2007;293:R528–R537.
Tamma G, Wiesner B, Furkert J et al. The prostaglandin E2 analogue sulprostone antagonizes vasopressin-induced antidiuresis through activation of Rho. J Cell Sci 2003;116:3285–3294.
Madsen KM, Clapp WL, Verlander JW . Structure and function of the inner medullary collecting duct. Kidney Int 1988;34:441–454.
Leviel F, Hübner CA, Houillier P et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010;120:1627–1635.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This study demonstrates a major role for the prostaglandin E2 (PGE2) EP1 receptor in mouse collecting duct transport and examines the PGE2/EP system in hypertensive mouse kidneys. The authors show that PGE2/EP1 attenuates arginine vasopressin-mediated water reabsorption in the inner medullary collecting duct and inhibits sodium reabsorption in the cortical collecting duct by epithelial sodium channels and pendrin-dependent pathways.
Rights and permissions
About this article

Cite this article
Nasrallah, R., Zimpelmann, J., Eckert, D. et al. PGE2 EP1 receptor inhibits vasopressin-dependent water reabsorption and sodium transport in mouse collecting duct. Lab Invest 98, 360–370 (2018). https://doi.org/10.1038/labinvest.2017.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.133
This article is cited by
-
The association of urinary prostaglandins with uric acid in hyperuricemia patients
BMC Nephrology (2022)
-
Prostaglandin E2 receptor EP1 (PGE2/EP1) deletion promotes glomerular podocyte and endothelial cell injury in hypertensive TTRhRen mice
Laboratory Investigation (2020)
-
Renal protective effect of nebivolol in rat models of acute renal injury: role of sodium glucose co-transporter 2
Pharmacological Reports (2020)
