
Abstract
The mechanisms by which the extreme desmoplasia observed in pancreatic tumors develops remain unknown and its role in pancreatic cancer progression is unsettled. Chemokines have a key role in the recruitment of a wide variety of cell types in health and disease. Transcript and protein profile analyses of human and murine cell lines and human tissue specimens revealed a consistent elevation in the receptors CCR10 and CXCR6, as well as their respective ligands CCL28 and CXCL16. Elevated ligand expression was restricted to tumor cells, whereas receptors were in both epithelial and stromal cells. Consistent with its regulation by inflammatory cytokines, CCL28 and CCR10, but not CXCL16 or CXCR6, were upregulated in human pancreatitis tissues. Cytokine stimulation of pancreatic cancer cells increased CCL28 secretion in epithelial tumor cells but not an immortalized activated human pancreatic stellate cell line (HPSC). Stellate cells exhibited dose- and receptor-dependent chemotaxis in response to CCL28. This functional response was not linked to changes in activation status as CCL28 had little impact on alpha smooth muscle actin levels or extracellular matrix deposition or alignment. Co-culture assays revealed CCL28-dependent chemotaxis of HPSC toward cancer but not normal pancreatic epithelial cells, consistent with stromal cells being a functional target for the epithelial-derived chemokine. These data together implicate the chemokine CCL28 in the inflammation-mediated recruitment of cancer-associated stellate cells into the pancreatic cancer parenchyma.
Similar content being viewed by others


Main
Among the top diagnosed malignancies in the United States, pancreatic cancer continues to have the poorest 5-year survival rate.1 The overwhelming majority of patients have unresectable disease and have limited options for chemotherapy. The standard of care for early stage, localized pancreatic cancer involves surgical resection, followed by adjuvant chemotherapy and/or radiation. Conventional treatment strategies include FOLFIRINOX, a highly toxic four-drug combination, or a combination of gemcitabine and nab-paclitaxel, which has also shown only incremental improvement in survival compared with gemcitabine alone.2, 3 However, the number of Food and Drug Administration approved options for pancreatic cancer is expanding to nearly a dozen various single chemotherapeutic drugs as well as a number of combination therapies.4, 5 Unfortunately, although progress has been made towards defining a more effective treatment regimen, patient prognosis is only extended in months when compared with the gemcitabine alone.6 This has provided the impetus for a plethora of pre-clinical studies over the last half-decade exploring novel strategies for treating pancreatic tumors. Based on initial studies demonstrating that the stroma is pro-tumorigenic, chemotherapies that target the pancreatic tumor stroma had shown initial promise.7, 8 However, more recent studies in animal models show that the tumor stroma may restrict pancreas cancer metastasis, suggesting that stroma-targeted therapies could exacerbate malignancy.9, 10 Targeting stromal–epithelial activity via sonic hedgehog inhibitors in a combination strategy has also led to mixed results in early-phase human clinical trials, with one trial showing worse outcomes compared with gemcitabine alone in metastatic pancreatic cancer, whereas a more recent trial using a maximal dosing strategy showed more promising antitumor activity in advanced solid tumors.11, 12 Clearly, additional work is needed to decipher the role of stroma, including desmoplasia and/or immune components, in the pathogenesis of ductal pancreatic adenocarcinoma.
The mechanisms directing the physical formation of the unique stroma within pancreatic tumors continue to be characterized. Sato et al13 first showed that increased cyclo-oxygenase-2 expression is shared between pancreatic tumor epithelial cells and fibroblasts. Since then, several studies have identified signaling factors that modulate pancreatic tumor fibroblast activation and survival in conjunction with pancreatic tumorigenesis.14, 15, 16 Much like the liver, the pancreas retains an organ-specific compartment of fibroblasts known as pancreatic stellate cells (PSCs).17 The cancer-associated fibroblasts that constitute the physical majority of the pancreatic tumor are currently hypothesized to be derived from several different cell types including quiescent resident PSCs, resident periductal and interlobular fibroblasts, mesenchymal bone marrow-derived myofibrocytes or epithelial cells that have undergone an epithelial–mesenchymal transition.18 During pancreatic inflammation and stress, PSCs can become activated from their quiescent state, lose their vitamin A and lipid stores, and enter a more prolific and increasingly motile state. Once activated, PSCs are capable of altering or remodeling the extracellular matrix (ECM) within the inflamed or damaged pancreas. Proinflammatory signaling cascades known to be altered in activated PSCs include NF-κB, JAK/STAT, and PPAR.16 In addition, activated PSCs secrete several factors such as transforming growth factor-β (TGF-β), the chemokine CXCL12, and plate-derived growth factor, which may then act on pancreatic tumor epithelial cells in a paracrine manner or on the activated PSCs themselves in an autocrine manner.19, 20, 21 These factors are implicated in dysregulated proliferation of the developing tumor epithelium and may also facilitate parenchymal invasion. Conversely, key epithelial produced factors that facilitate communication with activated PSCs/cancer-associated fibroblasts, coordinating stromal remodeling within the unique pancreatic tumor microenvironment, remain poorly defined.22
PSCs have an integral part in re-structuring the physical components of the tumor microenvironment.21 Implicit in these re-organizing events is the likely migration, recruitment, and spatial reorganization of PSCs to specific locations within the primary pancreatic tumor by malignant epithelial cells. Chemokines are a family of secreted proteins whose precise function is to facilitate directional movement of cells in a concentration-dependent manner. Although chemokines such as CXCL12 have been studied extensively in the context of cell death, proliferation, and cancer metastasis,23, 24, 25, 26, 27 the role that other chemokines have in fibroblast function, recruitment, or tumor ECM remodeling is comparatively unexplored. Herein, after completing a transcript expression screen of the 20 chemokine receptors in human pancreatic cancer cell lines, we tested the hypothesis that chemokines have an inflammation-driven role in the migration and recruitment of PSCs towards pancreatic tumor epithelial cells. We found increased expression of the chemokine ligands CCL28 and CXCL16 in human pancreatic cancer cell lines and tissue, relative to normal cells or pancreas. Their cognate receptors CCR10 and CXCR6, respectively, were also increased in expression. Notably, CCL28 and CXCL16 expression were localized primarily to cancer epithelial cells, whereas tumor-associated fibroblasts were characterized by a pronounced elevation in receptor expression. Further examination of human tumor specimens revealed that CCL28 expression was markedly upregulated during pancreatitis or following inflammatory cytokine signaling. CCL28 stimulation of three HPSC lines resulted in chemotactic migration as a functional response, without perturbing the activation state of the cells. These data support the notion that CCL28 secreted by pancreatic tumor epithelial cells direct the chemotactic recruitment of PSCs. Together, these data identify the novel involvement of the CCL28–CCR10 chemokine axis in pancreatic tumor epithelial–fibroblast interactions.
Materials and methods
Human Tissue Samples
De-identified human tissue specimens, including normal pancreas, pancreatitis, and pancreatic ductal adenocarcinoma (PDAC) tissue were provided in a blinded manner by the Surgical Oncology Biorepository of the Medical College of Wisconsin (MCW) with informed consent and in accordance with protocols approved by the MCW institutional review board. Normal pancreas tissue specimens were isolated exclusively from adjacent normal tissue discards of organ transplantation or pancreatic operations not involving PDAC or other exocrine malignancies. All the tissue sections were provided from whole-tissue paraffin-embedded blocks, rather than micro-arrays, to observe the surrounding microenvironment pathology. Disease status of those normal, inflamed, and tumor tissues was confirmed by a board-certified pathologist.
Immunohistochemistry
Immunohistochemical analysis was performed as previously published.27 Briefly, all the tissues were fixed, processed, embedded in paraffin, and then cut into 5-micron sections as needed by the Children’s Research Institute of the Children’s Hospital of Wisconsin histology core. Antigen retrieval was performed by boiling the tissue sections in 10 mM sodium citrate, followed by quenching in hydrogen peroxide, and antibody-specific blocking solution. The sections were incubated overnight in primary antibody against CXCR6 (Ab 8023 Abcam, Cambridge, UK), CXCL16 (Ab 101404 Abcam), CCR10 (Ab 30718 Abcam), CCL28 (MAB7171 R&D Systems, Minneapolis, MN, USA), CK19 (A53-B/A2 Abcam), smooth muscle actin (α-SMA, Ab7817 Abcam), and then HRP-conjugated secondary antibody (HRP-α-rabbit IgG (NA934) and HRP-α-mouse antibody (NXA931) GE Healthcare Life Sciences, Pittsburgh, PA, USA) the next day. Staining was visualized by the DAB chromogen (Vector Labs, Burlingame, CA, USA). Histochemical hematoxylin and eosin (H&E), Masson’s trichrome, and Movat’s pentachrome staining were performed by the histology core. Image acquisition and analysis of immunohistochemical stained tissue sections was completed using a Nikon Labphot 2 upright microscope. Intensity was assessed by a qualified histopathologist who was blinded to the tissue type (normal, pancreatitis, cancer) and antibody target. Epithelial duct and stroma staining intensity was graded 0=no staining, 1=weak staining, 2=moderate staining, and 3=strong staining relative to the epithelial marker CK19, or αSMA, the stromal cell marker.
Human and Murine Pancreatic Epithelial Cell Lines
The human pancreatic carcinoma cells Panc1, MiaPaCa2, Hs766T, Capan1, Capan2, and HPAFII were purchased from the American Type Culture Collection (ATCC, Rockville, MD USA) and cultured under the recommended conditions and as previously published.27 Human pancreatic epithelial nestin-expressing (HPNE) cells, an epithelial-like cell line used as a surrogate normal cell line, was purchased from ATCC and cultured as recommended. The patient-derived PDAC MCW#1, MCW#3 and MCW#4 cells were de-identified of patient identifiers and provided by the Surgical Oncology Biobank. In accordance with an IRB approved protocol and our prior publications,27, 28 patient-derived cells were isolated following manual and enzymatic digestion from consented patient specimens of primary pancreatic ductal adenocarcinomas, serially diluted and cultured in 2D tissue culture dishes in DMEM-F12 media containing 10% fetal bovine serum (FBS), penicillin/streptomycin (Pen/Strep), 100 U insulin (Sigma-Aldrich, Milwaukee, WI, USA), bovine pituitary extract (ThermoFisher Scientific, Carlsbad, CA, USA), epidermal growth factor (ThermoFisher) and hydrocortisone (Sigma). Unique cell lines of low passage (< passage 5) were analyzed. Patient-derived cells were positive for oncogenic KRas and p53 mutations. The cell lines were routinely validated using short tandem-repeat sequence analyses of 10 genetic loci, TPOX, D5S818, D16S539, 1vWA, TH01, D21S11, CSF1PO, D7S820, AMEL, and D13S317, to detect misidentified, cross-contaminated, or genetically drifted cells (Promega PowerPlex, Madison, WI, USA). The murine pancreatic cancer cell lines FC1199, FC1242, FC1245, and DT10022 were derived from founder KRasLSL.G12D/+-p53R172H/+-PdxCre (KPC) mice on a homogenous C57BL/6 background and were kindly provided by Dr Dannielle Engle and Dr David Tuveson (Cold Spring Harbor Laboratories, NY, USA). All KPC cells were isolated using standard enzymatic digestion and maintained in high glucose (4.5 g/l) DMEM with 10% (v/v) FBS plus pen/strep as recently described.7, 29
Human Pancreatic Fibroblast Cell Lines
The human pancreatic stellate cell line (HPSC) was derived from human PDAC tumor and immortalized with hTERT and SV40T plasmids under Neomycin antibiotic selection and used to model inflammatory cancer-associated fibroblasts as defined previously.14 The HPSC cell line was maintained in high glucose (4.5 g/l) DMEM with 10% (v/v) FBS plus pen/strep. Additional stellate cells, HPSC2 and HPSC3 cells, were obtained from fresh surgical human PDAC samples, characterized and immortalized with hTERT,30, 31 and maintained in high glucose (4.5 g/l) DMEM with 15% (v/v) FBS plus pen/strep.
Reverse Transcriptase-Polymerase Chain Reaction
RNA was isolated from cells and tissues using the RNAEasy kit from Qiagen (Germantown, MD, USA). All RNA samples were additionally treated with DNAse I to remove any genomic DNA contaminant before cDNA conversion. Conversion to cDNA was performed by priming with random hexamers and using the SuperScript II cDNA synthesis kit (ThermoFisher). The PCR products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining and ultraviolet imaging. The primers for the promoter region of the human mannose-binding lectin (MBL) or the mouse natural resistance-associated macrophage protein (NRAMP) genes were used to ascertain potential genomic DNA contaminant while amplification of the human GAPDH or mouse actin transcript was a loading control. Thirty cycles of amplification of chemokine receptors and ligand mRNA was done using validated oligonucleotide primer sets listed (Supplementary Table 1) or as published previously.23
Chemotaxis
Migration assays were conducted using 8 μm pore size Transwell inserts (Corning Costar, Corning, NY, USA). The insert membrane was coated with Collagen I (15 μg/ml) followed by blocking in 2% (w/v) BSA diluted in PBS. HPSC, HPSC2, or HPSC3 cells were grown to semi-confluence, serum starved for 2 h in the T75 flask before seeding, lifted using enzyme-free buffer (ThermoFisher), washed, and plated (105 cells) into the pretreated inserts. For the 2D co-culture assays, 2.5 × 105 Panc1, MiaPaCa2, or HPNE cells were seeded to 24-well dishes and serum starved 24 h before the assay. Stimulation or inhibition of migration was mediated via incubation (all treatments were added to the bottom well) with recombinant CCL28 (717-VC, R&D Systems) or polyclonal neutralizing antibodies against CCL28 (AF717, R&D Systems). Chemotactic migration was measured after 4 h and quantified from representative images taken using fluorescence microscopy after the upper sides of wells were swabbed, fixed, and stained with DAPI as done previously.27
Enzyme-Linked Immunosorbent Assay
Secreted chemokines were detected using a sandwich ELISA (hCCL28 DuoSet, R&D Systems). A total 105 cells were plated overnight in a 48-well plate and the next day culture medium switched to serum-free medium. Eighteen hours later, the cells were treated with 50 ng/ml interferon-gamma (IFNγ) for an additional 24 h. Supernatants were collected from control and IFNγ-treated cells and frozen at −80 °C before protein detection and quantification in accordance with the manufacturer’s directions. CCL28 was measured from a minimum of three biological replicates, with each sample assayed in triplicate.
Flow Cytometry
HPSCs were grown to 90% confluency, lifted with enzyme-free dissociation buffer and washed with PBS. The cells were counted and 1 × 106 cells aliquoted in 1% (w/v) BSA/PBS. From here on, all manipulations were on ice or at 4 °C. The cells were incubated for 1 h with CCR10 antibody (MAB3478 R&D Systems) or the isotype control (rat IgG, eBiosciences, San Diego, CA, USA). The cells were washed with BSA/PBS before and immediately after incubation with a chicken-anti-rat AlexaFluor 488 secondary antibody (Life Technologies, Madison, WI, USA). The cells were fixed in 1% (w/v) PFA in PBS and cell immunostaining data acquired using a BD-LSRII flow cytometer. The data were analyzed used FlowJo software (FLOWJO LLC, Ashland, OR, USA).
Extracellular Matrix Production by HPSCs
Extracellular matrix (ECMs) were obtained using an established fibroblast-derived ECM production protocol in an established in vivo-like 3D culturing system as published.31 Briefly, HSPCs were grown to confluency and treated with 50 μg/ml ascorbic acid for 8 days. The cells were stimulated daily with CCL28 (30 nM), TGF-β (10 ng/ml; C+) as a positive control, a TGF-β1R inhibitor SB-431542 (25 μM; C−), as a negative control, DMSO as a TGF-β/SB-431542 vehicle control (V), or left unstimulated (NS). The resultant unextracted 3D cultures were processed for phenotypic and quantitative intensity of αSMA characterization and ECM alignment analyses as published.30, 31, 32, 33
Indirect Immunofluorescence of Unextracted 3D HPSC Cultures
ECM-producing HPSC cultures were fixed, permeabilized, and processed as published.31 The samples were blocked using Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) containing 1% (v/v) donkey serum then incubated with either rabbit polyclonal anti-human fibronectin (1∶200) or 1A4 mouse anti-αSMA (1∶300), both from Sigma-Aldrich and diluted in blocking buffer. Fluorophore cross-linked secondary antibodies (Jackson ImmunoResearch Laboratories) were incubated at 1∶100 dilution. The samples were mounted using Prolong Gold anti-fading reagent from Life Technologies (Carlsbad, CA, USA) and scanned using a confocal spinning disk Ultraview (Perkin-Elmer Life Sciences, Boston, MA, USA) microscope. Images were acquired with a × 60 1.45 PlanApo TIRF oil immersion objective, under identical exposure conditions using Volocity 6.3.0 (Perkin-Elmer Life Sciences).
ECM Fiber Orientation Analysis
Monochromatic images corresponding to fibronectin fiber acquisitions were analyzed via ImageJ’s OrientationJ plugin (http://bigwww.epfl.ch/demo/orientation/) as described.31, 34 Image analysis outputs were normalized by identifying and setting mode angles to 0o and correcting angle spreads to fluctuate between −90° and 90°. Angle spreads for each experimental condition, corresponding to a minimum of three experimental repetitions and five image acquisitions per condition, were plotted and their standard deviations calculated using Excel. Percent of ECM fibers oriented between −15o and 15o, to denote fraction of fibers distributed near the 0° mode, was determined for each normalized image-obtained data.30, 31, 32, 33
Statistical Analyses
All statistical analyses were performed using GraphPad Prism 6. Paired analyses were calculated using a Student’s t-test, a Mann–Whitney, or log-rank test depending on the source of the data and assumed distribution. Multiple comparisons between groups were analyzed using a one-way ANOVA and either a Dunnett post hoc analysis to identify pair-wise differences between experimental and control groups or a Tukey post hoc analysis to identify pair-wise differences between distinct experimental groups. Statistical significance was defined as P≤0.05.
Results
Chemokine Expression Profile in Human Pancreatic Cancer Epithelial Cells
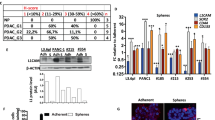
Our previous studies examining the role of chemokines in pancreatic cancer pathogenesis focused on the chemokine CXCL12 and its sole cognate receptor CXCR4.27, 29 However, as chemokines comprise a family of over 50 distinct ligands and 20 separate receptors, we next asked whether additional chemokines might be involved in pancreatic disease. Previous studies looking into chemokine expression in pancreatic cancer have been limited to specific ligands or receptors, including CXCR2 and CXCR4.27, 35, 36 Consequently, we profiled the chemokine receptor family in a battery of well-established human pancreatic cancer cell lines. Among CC-type chemokine receptors, we found the pronounced elevation in transcript for the Ccr10 gene in each of the cell lines (Figure 1a). Analysis of CXC-type receptors revealed that transcript for the Cxcr6 gene was robustly and uniformly expressed in each of the pancreatic cancer cell lines examined (Figure 1b). By contrast, none of the other 17 chemokine receptors had a similarly consistent or elevated expression pattern. We next investigated the expression of the cognate chemokine ligands for CCR10 and CXCR6. As shown in Figure 1c, the CCL27 and CCL28 ligands for CCR10 were routinely and strongly expressed in human pancreatic cancer cell lines. Notably, CCL27, the skin-specific ligand for CCR10 previously implicated in melanoma immune reactions,37 had weaker transcript expression compared with CCL28. Consistent with a prior report, CXCL16, the cognate ligand for CXCR6, was also expressed in each of the pancreatic cell lines (Figure 1d).38

Transcript analysis of the known chemokine receptors. (a, b) cDNA was isolated from a panel of human pancreatic cancer cell lines, Panc1, MiaPaCa2, Capan2, Capan1, HPAFII, and Hs766t, and screened by RT-PCR for expression of CCR (a), CXCR, CX3CR, or XCR (b) family of receptors. (c, d) The same panel of human cell lines was probed for expression of the known ligands for CCR10 (c) and CXCR6 (d). (e) RT-PCR analysis of patient-derived pancreatic cells (MCW PDAC cell lines) as well as a human pancreatic epithelial nestin-expressing (HPNE) cells confirmed CXCR6–CXCL16 and CCR10–CCL28 transcript expression. (f) Pancreatic tumor cells derived from the KPC murine model exhibited varying levels of the ligands and receptors. GAPDH and actin were analyzed as loading controls. Regions of the MBL and NRAMP genes were assessed as genomic DNA controls. Positive control was RNA from PBMCs.
We then broadened our examination of these two chemokine axes to include patient-derived pancreatic cancer cells, and contrasted expression in pancreatic cancer cells against the HPNE cell line isolated from nonmalignant pancreas. Just as shown in the established cell lines, each of the patient-derived lines expressed transcript for CCL28, CCR10, CXCL16, and CXCR6 (Figure 1e). Notably, in contrast to the tumor cell lines, the normal epithelial-like HPNE cell line lacked CCL28 mRNA and had minimal transcript expression for CXCL16. Given potential species-specific disparities in both chemokine biology and pancreatic cancer models, we further extended the analysis to include chemokine expression in an array of cell lines derived from an established genetically engineered murine model of pancreatic cancer. As shown in Figure 1f, RT-PCR analysis verified the robust and uniform transcript expression of CCL28–CCR10 and CXCL16–CXCR6 in mouse KPC pancreatic cancer cells.
Differential Expression of Chemokines in PDAC Epithelial and Stromal Compartments
Using immunohistochemistry techniques coupled with blinded scoring quantification, we next analyzed the expression of CCL28–CCR10 and CXCL16–CXCR6 in whole-tissue blocks from patients with both benign and malignant pancreas. We hypothesized that CCL28 and CXCL16 would be expressed in a pattern suggestive of a chemokine gradient for specific chemotaxis of cells within the tumor microenvironment. Compared with normal pancreatic ducts, we found elevated protein levels of CCL28 and CCR10 in PDAC epithelial cells (Figure 2a–d). Supplemental studies showed that CXCL16 and CXCR6 protein levels were increased in PDAC as well (Supplementary Figure 1). Parallel tissue sections immunostained with the ductal marker CK19 confirmed that these proteins were expressed largely within the cells of the ductal epithelium. Consistent with our previous observation of CXCR4, there was progressive elevation of CCL28–CCR10 (Figure 2c and d) and CXCL16–CXCR6 (Supplementary Figure 1) in precursor Pancreatic Intra-epithelial Neoplasms (PanIN). Interestingly, expression of CCR10 and CXCR6, but not their corresponding chemokine ligands, was also noted in the stromal compartment of PDAC tumors, as defined by immunostaining cancer-associated fibroblasts with an antibody against αSMA. Masson’s Trichrome or Movat’s Pentachrome staining of parallel tissue specimens was used to delimit and contrast the tumor stromal parenchyma from the epithelial compartment (Figure 2b–d; Supplementary Figure 1). Quantification of ligand and receptor staining intensity by an observer blinded to antibody or tissue stained indicated that CK19+ PDAC cancer cells are ligand and receptor positive, CCL28+/CCR10+, while αSMA+ stroma was ligand null and receptor positive, CCL28–CCR10+ (Figure 2d). Taken together, these mRNA and protein profile analyses reveal the consistent expression of CCR10 and its ligand CCL28 as a previously unrecognized chemokine receptor and chemokine in pancreatic cancer.

CCL28–CCR10 distribution in normal and malignant tissue. (a) Normal pancreatic tissue was sectioned and processed for histopathologic analysis for CCL28, CCR10, and CK19. The arrowhead denotes the epithelial cell lining. (b) Tissue from pancreatic tumor was sectioned and processed for immunohistochemical staining with antibodies specific for CCL28, CCR10, CK19, αSMA as well as H&E, Masson’s trichrome (3C) or Movat’s pentachrome (5C). ‘T’ denotes the tumor cells and ‘S’ denotes the stromal compartments of the tissue section as defined by CK19, αSMA and trichrome staining. (c, d) Representative stained tissue was scored by an investigator blinded to the tissue or antibody source for staining intensity. *P≤0.05, **P≤0.01, ***P≤0.001. Tissue staining shown in (a) and (b) representative of tissues sections from 14 normal, 12 PanIN, and 12 PDAC patients, based on clinical diagnosis and examination by a board-certified pathologist.
CCL28 Expression is Induced by Inflammatory Conditions
Chronic inflammation in the pancreas is a critical contributor to the generation of the requisite milieu for the formation of PDAC.39 Indeed, one of the common features between PDAC and pancreatitis, is the presence of activated stellate cells and the deposition of high levels of fibrotic materials.21, 40 We hypothesized that the expression of CXCL16 and CCL28 chemokine ligands may be triggered during inflammation of the pancreas. We performed immunohistochemical analyses on pancreatic tissue sections from patients diagnosed with pancreatitis and no clinical or pathological diagnosis of PDAC. Similar to prior reports of inflammation in the colon,41 CCL28 and CCR10 expression were elevated in both the stromal and epithelial compartments of pancreatitis tissue (Figure 3a–c). The increased levels of CCL28 in the stromal compartment in the inflamed, relative to the cancer, tissue, likely reflects its expression and production by epithelial or vascular cells present in pancreatitis and not in the immune suppressed cancer tissue. However, while CXCR6 expression was also elevated in pancreatitis epithelia and stroma, CXCL16 expression was significantly lower in both tissue compartments (Figure 3a–c). By comparison, examination of the chemokine ligand CXCL12 and its receptor CXCR4, showed that ductal epithelial cells in this nonmalignant tissue expressed the chemokine ligand CXCL12, with low levels of its cognate receptor CXCR4. The expression of CXCL12 and CXCR4 are similar to the levels observed in our prior work profiling normal pancreata, and agrees with its classification as a non-inducible chemokine axis refractory to inflammatory signaling27 (Figure 3d and e). These data suggest that pancreatic ductal CCL28 expression, in particular, may be driven by an inflammatory process. Cumulatively, the histopathological data suggest that the pronounced elevation of CCL28 by cancer cells, which is minimal in normal pancreas, creates a potential chemical gradient for stellate cell infiltration into the inflamed (pancreatitis), dysplastic (PanIN), and malignant (PDAC) tumor.

Immunohistochemical staining and scoring of pancreatitis tissue. (a) Sections of pancreatitis tissue were processed for immunostaining of CCR10 and CCL28, CXCR6, and CXCL16. (b, c) Scoring for the number of ductal (CK19+) or stromal (SMA+) regions for stain of CXCL16, CXCR6, CCL28, or CCR10. Gray line is the mean staining intensity of normal pancreatic tissue shown in Figure 2 and included as a reference. (d) Parallel tissue sections were processed for immunohistochemistry for CXCL12, CXCR7, CXCR4, and CK19. (e) Quantitative staining intensity of CXCL12, CXCR7, and CXCR4 in CK19+ cells. ***P≤0.001, ****P≤0.0001. Tissue staining shown in (a) and (d) are representative of tissue specimens from five to nine individual patients clinically and pathologically diagnosed with pancreatitis.
HPSC Chemokine Expression and Canonical Chemotactic Signaling
Previous work examining the functional role of the CCL28 chemokine ligand, and its receptor CCR10, in cancer have primarily focused on its impact on T-cell-mediated disease.37, 42 However, PDAC is notable for diminished levels of T-infiltrating lymphocytes relative to other cancers.43 Despite our observation that pancreatic fibroblasts express CCR10, there are no prior reports exploring the functional response of fibroblast cells to CCL28. To further test the hypothesis that elevated CCL28 expression in PDAC may be mediated by inflammatory signaling, we acquired the HPSC cell line, an activated stellate cell line previously isolated from human PDAC tissue and subsequently immortalized.14 Using a sandwich ELISA, we measured the level of CCL28 protein produced and secreted by an array of PDAC epithelial cell lines and HPSC cells. Three of the four cell lines produced basal quantities of CCL28 that could be increased following stimulation with IFNγ (Figure 4a). Notably, Capan2 cells produced little, if any, CCL28 before IFNγ treatment. In marked contrast, HPSC cells did not secrete any detectable CCL28 under either basal or inflammatory signaling induced conditions using IFNγ (Figure 4a). Flow cytometric analysis established the presence of surface levels of CCR10, the cognate receptor for CCL28, on HPSCs (Figure 4b). ELISA and flow cytometric analyses of the PDAC epithelial lines and the HPSC cells, respectively, confirmed that, together, the expression pattern of CCL28 and CCR10 mirrored the pattern observed in human tissues and validates their potential for use in relevant functional analyses. These data suggest tumor cells are a key source of CCL28 in the tumor microenvironment.

Functional characterization of the CCR10–CCL28 chemokine axis. (a) Sandwich ELISA revealed that the panel of tissue culture pancreatic cancer cells secrete CCL28 when stimulated with 50 ng/ml IFNγ. Levels of CCL28 were undetectable in HPSCs treated with IFNγ. (b) Flow cytometric analysis of HPSCs revealed the cell surface expression of CCR10 (top panel). The mean fluorescence intensity (MFI) was determined for cells stained with the isotype control primary antibody (light gray) and anti-CCR10 antibody (dark gray). (c) CCL28 stimulates chemotaxis. HPSCs were plated in the top well of a transwell insert in serum-free media. The bottom well contained media (serum free; SF or full growth; FG). Recombinant CCL28 (30 nM) was added to the bottom or top well as indicated. After 4 h, inserts were swabbed, stained with DAPI, and enumerated. (d) CCL28-mediated HPSC migration is dose-dependent. HPSCs were plated in the top well of a transwell insert. The bottom well contained either full-growth media (+; positive control) or serum-free media that either lacked stimulant (−; negative control), or increasing concentrations of recombinant CCL28. (e) HPSC migration is mitigated by an anti-CCL28 neutralizing antibody. The transwell migration setup as above was modified to include the following conditions: no stimulation (NS; negative control), 10 ng/ml TGF-β (positive control) or 30 nM recombinant CCL28. Neutralizing antibody to CCL28 was added to the bottom well for 30 min before incubation with the cells. (f) CCL28-mediated directional migration of HPSCs is through a G-coupled protein receptor pathway. HPSCs were pretreated with pertussis toxin (PTX) or vehicle, before CCL28 stimulation. (g) Transcript expression of CCR10 in HPSC cells. RNA was isolated and used as a template for RT-PCR analysis using CCR10 or GAPDH as a loading control. (h) CCL28 stimulates chemotactic migration of patient-derived HSPC2 cells. HPSC2 cells were added to a transwell insert and placed in a well containing either serum-free medium in the absence (NS) or presence of CCL28 (CCL28) or full-growth medium (FG) as a positive control (FG). *P ≤ 0.05, **P ≤ 0.01, ***P≤0.001.
We next sought to characterize the ability of HPSC cells to functionally respond to CCL28 stimulation via canonical chemokine signaling. Chemotactic migration typically occurs through a Gαi-protein-mediated process, as has been shown previously with CCR10-expressing T cells.44 As predicted, CCL28 directed the dose- and receptor-dependent chemotactic migration of HPSCs (Figure 4c and d). HPSC migration was prevented when equal concentrations of ligand were added to top and bottom chambers (Figure 4c) or using a ligand neutralizing antibody (Figure 4e, gray bars). CCL28-mediated HPSC migration was abrogated using pertussis toxin, a specific inhibitor of Gαi-protein signaling (Figure 4f). Live cell counting over 72 h showed that the CCL28 chemokine ligand did not stimulate significant changes in population growth or death (not shown). To determine whether the effect of CCL28 was more broadly applicable to human pancreatic cancer stellate cells, additional patient-derived HPSCs were screened for responsiveness to this chemokine. Consistent with the HPSC cell line, patient-derived HPSC2 and HPSC3 cell lines similarly expressed CCR10 (Figure 4g). Further, CCL28 stimulated a comparable level of chemotactic migration in both HPSC and HPSC2 cell lines (Figure 4h). Cumulatively, these data are the first to indicate that CCL28 stimulated chemotaxis of PDAC-associated stellate cells through canonical chemokine receptor signaling.
Activation Status of Stellate Cells is Not Altered by CCL28 Treatment
To determine whether CCL28 treatment modified the activation of HPSCs, we evaluated three individual PDAC-associated HPSCs14, 30, 31, 45 for the expression of αSMA and production of anisotropic (ie, aligned) ECM.31, 32, 33 The cells were treated with CCL28 or TGF-β as a positive control (C+). Treatment with the TGF-βR1 inhibitor SB-431542 (C−), a vehicle (V) or cell left untreated, were three negative controls. Representative immunofluorescence images, shown in Figure 5a, indicated that each of the three HPSC cell lines responded similarly to increasing (C+) and decreasing (C−) TGF-β activity by altering αSMA levels. Semi-quantitative image analysis, shown in Figure 5b, verified that αSMA levels were significantly elevated following treatment with the TGF-β positive control (C+) or were diminished when treated with the SB-431542 negative control (C−). By contrast, αSMA levels were largely unchanged in each of the HPSC lines following daily incubation with CCL28.

CCL28 does not alter activated phenotype of HPSCs. Three independent HPSC lines were treated with 30 nM CCL28, left untreated (NS), treated with 10 ng/ml TGF-β (C+), treated with SB-431542 (25 μM), a TGF-β1R inhibitor (C−) or treated with vehicle controls (V). (a) Representative immunofluorescence images show digital intensity ‘heat maps’ of detected αSMA, corresponding to intensity color bar scale. Scale bars represent 50 μm. (b) Semi-quantitative imaging analysis of the mean integrated intensity (Integ Int) of αSMA detection normalized for each HPSC line (HPSC, 2, 3) using the vehicle (V) set as 1 arbitrary unit. HPSC: #P=0.0179, ***P≤0.0002; HPSC2: #P=0.0144, ****P<0.0001; HPSC3: ****P<0.0001. (c) Representative images of HPSC-derived ECMs produced under experimental conditions as in (a) and analyzed using OrientationJ plugin of ImageJ software. Color tones were normalized using hue values for common mode angle (cyan/green boarded color) visualization as represented on the orientation bar. (d) Box and whisker plot of mean percent of fibers distributed within 15° angles from the mode corresponding to the indicated experimental conditions. HPSC: #P=0.0304, *P=0.028; HPSC2: *P=0.0117; HPSC3: C+ versus V, *P=0.0101. No substantial ECM production was obtained from HPSC3 under TGF-β inhibition and was designated as not available (NA).
Next, we investigated whether CCL28 stimulation had any appreciable effects on ECM production. Again, each of the three HPSC lines were plated and analyzed for fibronectin fiber alignment via ImageJ analysis. As highlighted in Figure 5c and d, ECMs produced in the presence or absence of TGF-β (C+ and V, respectively) remain anisotropic (ie, organized) in activated HPSCs, while ECMs are rendered isotropic (ie, disorganized), or their formation is prevented altogether, under TGF-β blockade (C−). In contrast, no significant changes in ECM fiber production or organization were noted when the various HPSCs were stimulated with CCL28, compared with vehicle controls (Figure 5c and d). Cumulatively, these data demonstrate that CCL28 chemokine stimulation of resting HPSCs had little, if any, effect on the activation state of pancreatic cancer-associated stellate cells.
PDAC Epithelial Cells Attract Fibroblasts Through CCL28 Secretion
Given the CCL28–CCR10 expression profile and our subsequent functional analysis of HPSCs, we hypothesized that CCL28 produced by tumor cells (shown in Figure 4a) directed the chemotactic migration of CCR10-expressing stellate cells within the pancreatic tumor microenvironment. To test this hypothesis, we constructed a 2D co-culture transwell assay to model intercellular communication within the tumor microenvironment. This approach has been successfully used in previous studies to model endothelial–immune cell interactions.46 In this model, PDAC epithelial cells cultured in the bottom chamber were stimulated 24 h with IFNγ to produce a chemokine ‘source’ gradient. The ‘recipient’ HPSCs were then added to the top chamber of the transwell and the number of migrating activated fibroblasts enumerated 4h later (Figure 6a). Using this assay, we found that HPSCs chemotactically migrated across the membrane insert towards Panc1 cancer cells only after those source cells had been stimulated with IFNγ (Figure 6b and c). Notably, HPSC migration towards Panc1 cells was completely abolished by neutralizing anti-CCL28 antibody (Figure 6b and c). Moreover, CCL28-dependent chemotactic migration of HPSCs was also observed toward MiaPaCa2 cells as a separate PDAC epithelial source, as shown in Figure 6d and e. Last, chemotactic migration was specifically toward transformed tumor cells such as Panc1 and not HPNE cells, an epithelial-like cell used as a surrogate normal exocrine pancreatic epithelium (Figure 6f and g). In total, we have shown that pancreatic cancer cells direct pancreatic cancer-associated stellate cell migration through the pathologic elevation in CCL28 production.

Pancreatic tumor cells secrete CCL28 and promote HPSC migration. Schematic representation of the experimental plan (a). Briefly, PDAC epithelial cells were seeded to the bottom well of a transwell dish (1) then stimulated with 50 ng/ml IFNγ to elicit chemokine secretion. HPSCs were plated onto the top chamber of the transwell insert (2) while CCL28 neutralizing antibody was added to the bottom chamber. The insert with HPSCs was added (3) and incubated for 4h before (4) fixing, DAPI-staining, and cell counting. Stimulation of Panc1 (b, c) and MiaPaCa2 (d, e) cells by IFNγ resulted in directional migration of HPSCs that was inhibited by the neutralizing CCL28 antibody. HPSC migrate towards PDAC tumor cells and not non-transformed epithelial HPNE cells (f, g). Representative images (c, e) of five independent biological replicates are presented. *P≤0.05, **P≤0.01.
Discussion
Chemokines comprise a large group of secreted proteins that influence the pathophysiology of a variety of disease states. Though chemokines have been extensively studied in several inflammatory conditions and cancers,47 there has yet to be an expansive analysis of the role that chemokines have in the development of pancreatic cancer and other pancreatic diseases. Functional studies of chemokines in pancreatic cancer have primarily been limited to the CXCL12–CXCR4 chemokine axis.27, 35 Our study aimed to more broadly understand the expression patterns of the chemokine receptor family in pancreatic disease and functional roles in pancreatic cancer. Through analysis of both human cell lines and patient-derived tissue, we uncovered a chemokine ligand-receptor expression pattern suggestive of a novel chemokine gradient involved in the directed migration, as opposed to activation, of activated stellate cells and/or mesenchymal stem cells into the pancreatitis inflamed and pancreatic cancer microenvironments.
Broadly, our study is the first to implicate a role for the CCL28–CCR10 chemokine axis in pancreatic disease. Our initial transcript analysis revealed that the chemokine receptors CCR10 and CXCR6, along with their cognate respective chemokine ligands, CCL28 and CXCL16, were expressed in nine human and four mouse pancreatic cancer cell lines. The changes in expression of CXCL16 and CXCR6 observed in pancreatitis and pancreatic cancer specimens mirrored those reported previously.38 Although Wente et al38 did not specifically examine the role of CXCL16 in the stroma, and rather focused on the autocrine role of CXCL16 in PDAC epithelial proliferation, we initially focused our analysis on CCL28 and CCR10. Subsequent immunohistochemical and fluorescent staining of normal pancreata and patient-derived PDAC tissue not only demonstrated the upregulation of both sets of chemokine receptors and ligands in diseased epithelium compared to normal ducts, but also uncovered differential expression pattern of CCL28 and CCR10 in epithelial and stromal compartments with expression of the chemokine restricted to the CK19 positive ductal cells. Although prior studies have reported on the role of CCL28 and CCR10 in T-cell-mediated inflammation in colitis, ovarian cancer, and skin cancer and dermal mesenchymal stem cell trafficking,37, 41, 42 neither the ligand nor the receptor have previously been implicated in expression studies of fibroblasts. These analyses reveal a potentially biologically significant difference in chemokine expression in tumor epithelial cells, as well as fibroblasts. In total, these data suggested a potential paracrine role for CCL28 and/or CXCL16 within the context of the larger pancreatic tumor microenvironment.
Prior work looking into the specific factors that govern the formation of the pancreatic tumor microenvironment have been primarily limited to the study of Hedgehog signaling, which stimulates growth and proliferation of fibroblasts.19 Others have shown that the pancreas is a repository for quiescent fibroblasts known as stellate cells.17 In either the progression to malignancy or under chronic inflammatory conditions, these cells become activated and reorganize to facilitate the deposition of fibrotic materials that typify the extreme desmoplasia observed in pancreatic tumors. Our analysis of patient-derived pancreatic cancer-associated stellate cells demonstrated that CCL28 does not affect levels of activation or ECM deposition traits. Moreover, when looking at patient-derived nonmalignant pancreatitis tissue, we observed that elevated CCL28 expression occurs in patients with chronic inflammation of the pancreas. Elevated levels of CCL28 in the inflamed stromal compartment likely reflects its expression by vascular endothelial cells as our culture data indicated CCL28 was not increased by IFNγ treatment. This suggested that inflammatory conditions are a potential driving factor for CCL28 expression in the tumor microenvironment. Indeed, treatment of a panel of cultured PDAC epithelial cell lines and HSPCs with the inflammatory cytokine IFNγ confirmed that CCL28 is secreted by epithelial cells, but not fibroblasts. These data parallel prior reports regarding the regulation of CCL28 expression under inflammatory conditions.41, 44 However, others have suggested that expression of CCL28 can be induced by tumor hypoxia,42 which is a hallmark of PDAC. We believe this is the first report analyzing human pancreatitis tissue that shows elevated levels of the chemokine CCL28 specifically by epithelial and stromal cells in the inflamed pancreas. This expression pattern suggests that CCL28 may have a role in the evolution of pancreatic tumor microenvironment.
Despite the wealth of studies examining the role of fibroblasts in PDAC pathogenesis, particularly in the context of tumor proliferation and drug-delivery,7, 8 no previous studies have probed the mechanism directing the migration of stellate cells or fibroblasts in the tumor. Given that chemotaxis occurs in a biphasic manner, with lower levels of chemokine stimulating migration and higher levels arresting migration, our expression analysis of CCL28 suggested the presence of a potential chemotactic gradient within pancreatic tumors that permits the migration of fibroblasts towards tumor epithelia. Using the already established HPSC cell line as a model for pancreatic tumor fibroblasts,14 we first showed that HPSCs mimic the expression pattern of CCL28 and CCR10 observed in pancreatic tissue. Thus, these stellate cells produce little, if any chemokine, but robustly express functional CCR10 receptor. We then verified that HPSC cells chemotactically migrated towards exogenous CCL28 protein in a transwell migration assay. Importantly, HPSC cells functionally responded to CCL28 in a G-protein signaling-dependent manner. The HPSC migration response to CCL28 was independent of change in cell viability. Finally, to test the hypothesis that a chemotactic gradient exists between PDAC epithelial cells and fibroblasts, we used a 2D co-culture transwell assay and found that HPSC cells chemotactically migrated towards pancreatic cancer epithelial cells that had been pretreated with IFNγ. HPSC migration towards PDAC epithelial cells was reversible with a neutralizing antibody for the CCL28 protein. In sum, these data support a model wherein CCL28 has a functional role in the recruitment of pancreatic stellate cells in the PDAC stroma.
On the basis of our results, we propose a mechanistic model in which a chemokine gradient develops in parallel with pancreatic disease progression (Figure 7). Thus, normal ducts show the presence of CCR10, but no CCL28, whereas quiescent stellate cells are deficient in both ligand and receptor (Figure 7a). Through inflammation, stellate cells become activated and ductal epithelial cells are stimulated to secrete CCL28, which then permits the stellate cells to migrate in a directed manner towards the epithelia (Figure 7b). In PDAC tumorigenesis, this pathologic CCL28 gradient, established by epithelial cancer cells, directs the localization of PDAC fibroblasts or mesenchymal stem cells into close proximity and facilitates a desmoplastic reaction (Figure 7c). Cumulatively, this provides a novel mechanism for the recruitment of fibroblasts into the pancreatic cancer stroma. Further, in a tumor microenvironment with relatively few T cells, the model identifies a novel role for CCL28, which has yet to be associated with directing chemotaxis of non-immune cells.

Schematic model depicting the functional role for CCL28–CCR10 chemokine signaling between pancreatic cancer cells and activated stellate cells. Normal pancreatic duct epithelial cells and quiescent stromal fibroblasts almost no ligand CCL28 (purple circles) and basal levels of the receptor CCR10 (a). In inflammation, such as in pancreatitis, CCL28 expression is increased by cytokines such as IFNγ, which also participate in the activation of stellate cells into cancer-associated fibroblasts. CCL28 directs the migration of stellate cells toward the epithelium (b). CCL28 produced by transformed ductal cells direct the sustained migration of activated stellate cells into the remodeling tumor microenvironment (c).
Moving forward, these data add to the burgeoning body of literature concerning the pancreatic cancer stroma. Although our study does not address whether CCL28 is pro- or anti-malignant, targeting PDAC–stroma interactions through the CCL28–CCR10 axis may provide a novel avenue for modulating the dynamics, as opposed to the activation of PDAC tumor stroma. For example, enhancing tumoral CCL28 may compound the inflammatory fibrosis in PDAC patients that is reported to influence PDAC malignancy and metastasis.10 The cumulative work in this study identifies CCL28 as a unique tumoral factor that helps to regulate dynamics of pancreatic cancer desmoplasia and merits additional investigation into the CCL28–CCR10 chemokine axis in future studies.
References
Siegel R, Naishadham D, Jemal A . Cancer statistics, 2013. CA Cancer J Clin 2012;62:10–29.
Von Hoff DD, Ervin T, Arena FP et al, Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–1703.
Conroy T, Desseigne F, Ychou M et al, FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–1825.
El-Hadaad HA, Wahba HA . Oxaliplatin plus 5-fluorouracil and folinic acid (OFF) in gemcitabine-pretreated advanced pancreatic cancer: a phase II study. J Gastrointest Cancer 2013;44:313–317.
Postlewait LM, Ethun CG, Kooby DA et al, Combination gemcitabine/cisplatin therapy and ERCC1 expression for resected pancreatic adenocarcinoma: results of a phase II prospective trial. J Surg Oncol 2016;114:336–341.
Burris HA, Moore MJ, Andersen J et al, Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–2413.
Olive KP, Jacobetz MA, Davidson CJ et al, Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009;324:1457–1461.
Provenzano P, Cuevas C, Chang A et al, Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–429.
Rhim A, Oberstein P, Thomas D et al, Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–747.
Özdemir B, Pentcheva-Hoang T, Carstens J et al, Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–734.
Kim EJ, Sahai V, Abel EV et al, Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res 2014;20:5937–5945.
Goldman J, Eckhardt SG, Borad MJ et al, Phase I dose-escalation trial of the oral investigational hedgehog signaling pathway inhibitor TAK-441 in patients with advanced solid tumors. Clin Cancer Res 2015;21:1002–1009.
Sato N, Maehara N, Goggins M . Gene expression profiling of tumor–stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res 2004;64:6950–6956.
Hwang RF, Moore T, Arumugam T et al, Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008;68:918–926.
Masamune A, Kikuta K, Watanabe T et al, Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol 2008;295:G709–G717.
Masamune A, Kikuta K, Watanabe T et al, Fibrinogen induces cytokine and collagen production in pancreatic stellate cells. Gut 2009;58:550–559.
Apte MV, Haber PS, Applegate TL et al, Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43:128–133.
Nielsen MFB, Mortensen MB, Detlefsen S . Key players in pancreatic cancer-stroma interaction: cancer-associated fibroblasts, endothelial and inflammatory cells. World J Gastroenterol 2016;22:2678–2700.
Bailey JM, Swanson BJ, Hamada T et al, Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res 2008;14:5995–6004.
Gao Z, Wang X, Wu K et al, Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology 2010;10:186–193.
Apte M, Pirola R, Wilson J . Pancreatic stellate cells : a starring role in normal and diseased pancreas. Front Physiol 2012;3:344.
Tape CJ, Ling S, Dimitriadi M et al, Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 2016;165:910–920.
Wendt MK, Johanesen PA, Kang-Decker N et al, Silencing of epithelial CXCL12 expression by DNA hypermethylation promotes colonic carcinoma metastasis. Oncogene 2006;25:4986–4997.
Wendt MK, Cooper AN, Dwinell MB . Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene 2008;27:1461–1471.
Ramos EA, Camargo AA, Braun K et al, Simultaneous CXCL12 and ESR1 CpG island hypermethylation correlates with poor prognosis in sporadic breast cancer. BMC Cancer 2010;10:23.
Baumhoer D, Smida J, Zillmer S et al, Strong expression of CXCL12 is associated with a favorable outcome in osteosarcoma. Mod Pathol 2012;25:522–528.
Roy I, Zimmerman NP, Mackinnon AC et al, CXCL12 chemokine expression suppresses human pancreatic cancer growth and metastasis. PLoS One 2014;9:e90400.
Kim MP, Evans DB, Wang H et al, Generation of orthotopic and heterotopic human pancreatic cancer xenografts in immunodeficient mice. Nat Protoc 2009;4:1670–1680.
Roy I, McAllister DM, Gorse E et al, Pancreatic cancer cell migration and metastasis is regulated by chemokine-biased agonism and bioenergetic signaling. Cancer Res 2015;75:3529–3542.
Lee HO, Mullins SR, Franco-Barraza J et al, FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011;11:245 2407-11-245.
Franco-Barraza J, Beacham DA, Amatangelo MD et al, Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr Protoc Cell Biol 2016;71:10.9.1–10.9.34.
Amatangelo MD, Bassi DE, Klein-Szanto AJ et al, Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol 2005;167:475–488.
Goetz JG, Minguet S, Navarro-Lerida I et al, Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011;146:148–163.
Rezakhaniha R, Agianniotis A, Schrauwen JT et al, Experimental investigation of collagen waviness and orientation in the arterial adventitia using confocal laser scanning microscopy. Biomech Model Mechanobiol 2012;11:461–473.
Billadeau DD, Chatterjee S, Bramati P et al, Characterization of the CXCR4 signaling in pancreatic cancer cells. Int J Gastrointest Cancer 2006;37:110–119.
Ijichi H, Chytil A, Gorska AE et al, Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 2011;121:4106–4117.
Murakami T, Cardones AR, Finkelstein SE et al, Immune evasion by murine melanoma mediated through CC chemokine receptor-10. J Exp Med 2003;198:1337–1347.
Wente MN, Gaida MM, Mayer C et al, Expression and potential function of the CXC chemokine CXCL16 in pancreatic ductal adenocarcinoma. Int J Oncol 2008;33:297–308.
Guerra C, Schuhmacher AJ, Cañamero M et al, Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-ras oncogenes in adult mice. Cancer Cell 2007;11:291–302.
Walter K, Omura N, Hong S et al, Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther 2008;7:882–888.
Ogawa H, Iimura M, Eckmann L et al, Regulated production of the chemokine CCL28 in human colon epithelium. Am J Physiol Gastrointest Liver Physiol 2004;287:G1062–G1069.
Facciabene A, Peng X, Hagemann IS et al, Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and treg cells. Nature 2011;475:226–230.
Bayne L, Beatty G, Jhala N et al, Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012;21:822–835.
Wang W, Soto H, Oldham ER et al, Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J Biol Chem 2000;275:22313–22323.
Goicoechea SM, Bednarski B, Stack C et al, Isoform-specific upregulation of palladin in human and murine pancreas tumors. PLoS One 2010;5:e10347.
Mul FPJ, Zuurbier AEM, Janssen H et al, Sequential migration of neutrophils across monolayers of endothelial and epithelial cells. J Leukoc Biol 2000;68:529–537.
Roy I, Evans DB, Dwinell MB . Chemokines and chemokine receptors: update on utility and challenges for the clinician. Surgery 2014;155:961–973.
Acknowledgements
We thank Donna McAllister for expert technical assistance and authentication and validation of the cell lines. The work was supported, in part, by a grant from the MCW Cancer Center and continuing philanthropic donations from the Bobbie Nick Voss Charitable Foundation (to MBD). IR is a member of the Medical Scientist Training Program at MCW, which is partially supported by an NIGMS training grant (T32GM080202). This study was also partially supported by DOD’s W81XH-15-1-0170, NCI R01 CA113451 (to EC), and R01 CA113451-10S1 (to JF-B and EC), as well as NCI CA06927 support of the Talbot Library and Tissue Culture facility at Fox Chase Cancer Center. Finally, we would like to recognize funds from the Commonwealth of Pennsylvania and kind gift from Mrs Greenberg (to EC), in support of Fox Chase Cancer Center’s Pancreatic Cancer Initiative Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MBD has ownership interests in Protein Foundry, LLC. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Recent reports implicate stromal remodeling by stellate cells in pancreatic cancer with either pro- or anti-tumorigenic roles. The mechanisms coordinating stellate cell recruitment and activation remain enigmatic. This study indicates that inflammation-directed production of tumoral CCL28 influences recruitment and spatial reorganization of stromal remodeling by stellate cells in pancreatic cancer.
Supplementary information
Rights and permissions
About this article

Cite this article
Roy, I., Boyle, K., Vonderhaar, E. et al. Cancer cell chemokines direct chemotaxis of activated stellate cells in pancreatic ductal adenocarcinoma. Lab Invest 97, 302–317 (2017). https://doi.org/10.1038/labinvest.2016.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.146
This article is cited by
-
Targeted biologic inhibition of both tumor cell-intrinsic and intercellular CLPTM1L/CRR9-mediated chemotherapeutic drug resistance
npj Precision Oncology (2021)
-
Glycometabolic rearrangements--aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications
Journal of Experimental & Clinical Cancer Research (2020)
-
STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models
Journal for ImmunoTherapy of Cancer (2019)
-
The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma
Molecular Cancer (2018)
-
Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models
BMC Cancer (2018)
