
Abstract
Despite tremendous progress in the last decade, lung adenocarcinoma still represents a tumor with unfavorable prognosis when detected at advanced clinical stage. High-stage tumors are not amenable to surgical resection, and therefore systemic therapies are needed to control these tumors to prolong patient survival. In the era of molecular and personalized therapeutics, the discovery of mutations in epidermal growth factor receptor (EGFR) in 15–20% of lung adenocarcinomas and the associated response to EGFR-targeting tyrosine kinase (TK) inhibitors have provided a successful avenue of attack in high-stage adenocarcinomas. In this review, we will provide an overview of the EGFR pathway, review the significant somatic EGFR alterations in lung adenocarcinoma and highlight their implications for treatment. In addition, we will examine pathways by which tumors resist EGFR TK therapy, both as primary nonresponders and by acquired resistance. In doing so, we will examine other oncogenic pathways whose status in tumor samples may impact therapeutic responses despite presence of activating EGFR mutations.
Similar content being viewed by others

Main
Although prostate cancer and breast cancer remain the most common malignancies, malignant lung tumors are the leading cause of death in adult men and women.1 Broadly, malignant lung tumors can be subdivided as small-cell lung carcinoma (SCLC) and non-SCLC (NSCLC), with adenocarcinoma as the most frequently encountered histology, within NSCLC, as well as overall rate. Although the treatment for early-stage lung adenocarcinomas is primarily surgical, high-stage tumors usually are treated with chemotherapy with overall survival of <2 years.2 As the advent of lung cancer screening in selected populations is a fairly recent occurrence, a significant proportion of patients will be diagnosed with late-stage lung cancer.
A possible solution to this ongoing problem is the identification and unraveling of novel signaling pathways on which cancer cells selectively depend for growth and survival, setting the stage for cancer-specific treatment without the occurrence of unwanted side effects. Such advances occurred in chronic myelogenous leukemia and gastrointestinal stromal tumors in which tyrosine kinase (TK) inhibitors targeting the ABL kinase and the KIT kinase resulted in relatively selective cell death in these neoplasms. Reminiscent of the bcr-abl fusion gene in chronic myelogenous leukemia and KIT mutations in gastrointestinal stromal tumors and their response to imatinib,3 lung adenocarcinomas were found to harbor epidermal growth factor receptor (EGFR) mutations, also a cell-surface protein with intracellular TK activity. These targetable activating mutations render tumors susceptible to the drugs gefitinib and erlotinib.
EGFR PROTEIN AND ITS FUNCTIONAL PATHWAYS
In several malignancies, including lung adenocarcinomas, ductal carcinomas of the breast, and glioblastoma, ErbB receptor signaling is deregulated, driving uncontrolled proliferation of tumor cells, conferring the ability to evade programmed cell death, enhancing their ability to migrate, and facilitating metastasis.
EGFR is a 486 amino-acid receptor protein of 170 kDa with a single transmembrane sequence between 4 extracellular and 3 intracellular domains. Ligand-binding activity is in the extracellular domain 3; EGFR ligands include EGF, transforming growth factor-alpha, and amphiregulin.4 The binding of EGFR ligands induces dimerization allowing for receptor phosphorylation; such dimers can be homodimers or heterodimers with other HER family receptors.5 Several members that belong to the ErbB family have been described: HER1 (EGFR/erbB1), HER2 (neu, erbB2), HER3 (erbB3), and HER4 (erbB4).6 These different receptors display a specific distribution and molecular alteration pattern depending on the tumor entity. For instance, HER2 reveals an increased membranous expression in about one-third of breast carcinomas, predicting their susceptibility to trastuzumab.
The intracellular component of the EGFR protein includes a juxtamembrane domain, a region housing the TK activity, and a C-terminal domain. The kinase activity is dependent on a lysine residue at amino acid 721, but key components affecting ATP affinity are found within that domain. Alterations in the juxtamembrane domain and C-terminal domains can influence ligand binding. More importantly, numerous tyrosine residues are present in the C-terminal domain, and phosphorylation in these sites influence protein–protein interactions that are essential for signal transduction via pathways such as those mediated by PI3K and rasGAP/KRAS.5
Although each domain has specific function, truncation of exons 2–7 can cause a cell lineage-specific activation, which can also be associated with amplification. This alteration was described in 1985 in gliomas, and for many years this was the best demonstration of the oncogenic potential of constitutive EGFR activation.7 This alteration affects receptor recycling and results in constitutive activity that is independent of ligand binding.8
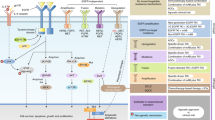
Dimerization of EGFR leads to TK activity and receptor phosphorylation. This phosphorylation induces interaction with GRB2, then SOS that leads to RAS activation. What follows is a cascade of kinase activations including BRAF and MEK1, ultimately resulting in ERK1/2 phosphorylation and activation.9 This signaling pathway has effects on cellular proliferation. In addition, the PI3K pathway is activated, causing an increase of its downstream effector protein kinase-B, also referred as AKT.10 Dephosphorylation of AKT is regulated by PTEN, a pivotal phosphatase with tumor-suppressive properties. PTEN is known to be mutated (for example, homozygous deletions) or epigenetically silenced in several malignancies, including glioblastoma and uterine endometrioid adenocarcinoma. PTEN possesses also the capability to mediate activation status of EGFR by influencing EGFR phosphorylation status.11 AKT itself regulates apoptotic signaling by phosphorylating and driving the expression of certain anti-apoptotic molecules, such as the inhibitor of apoptosis protein survivin12 (Figure 1).

Epidermal growth factor receptor (EGFR) pathway. The figure emphasizes on the elements of the signal transduction pathway for EGFR that are known points of mutation in lung adenocarcinoma. Provided are the relevant sections of text in which alterations in the particular pathway element is discussed.
An additional important downstream target is the mTOR pathway that affects protein translation, and is implicated in cell cycle progression, apoptosis, and metastasis. Being often constitutively active in several highly treatment refractory cancers, the mTOR pathway has also emerged as a target in cancer therapy. STAT signaling is also affected by EGFR signaling. For instance, STAT3 is phosphorylated and activated by activating EGFR mutations in lung adenocarcinomas.13
PRIMARY EGFR MUTATIONS AND TK INHIBITOR THERAPY
Activating mutations in oncogenes have been described as an ‘Achilles heel’ in cancer, coining a term of ‘oncogene addiction’.14 Dependency of a tumor on a specific oncogene renders these malignancies potentially sensitive to inhibitors that preferentially target the altered oncogene and thereby abrogates the tumor-promoting properties of it. In fact, sudden cessation of signaling can induce cell death.
Targeting EGFR in lung cancer began before knowledge of the existence of specific activating mutations. Gefitinib, when used in unselected populations, still resulted in a response rate of 10–20%.15, 16 Erlotinib administration was associated with an improvement in overall survival, but this important finding was overshadowed by more dramatic results in certain subgroups—adenocarcinoma histology, female gender, nonsmoking status, and Asian ethnicity.17 This finding of patients with lung adenocarcinomas with response to gefitinib and erlotinib (EGFR TK inhibitors or EGFR TKI) led to molecular investigation of the EGFR pathway in these tumors.
In studies published in 2004, several mutations that render lung adenocarcinomas sensitive to gefitinib were identified.18, 19 The identification of these mutations was based on the observation that ∼10% of patients with NSCLC were responsive to gefitinib,18 which paralleled the finding that ∼8% of untreated patients had tumors with EGFR mutations. The fraction of patients who were sensitive to gefitinib were subjected to mutation analysis of the EGFR gene18 and eight of nine patients showed a mutation in the ATP-dependent TK domain (‘catalytic kinase domain’) in the EGFR gene.18 Most of these mutations were point mutations or in-frame deletions, which were present in a heterozygous pattern.18 Specifically, they found three point mutations (G719C, L858R, and L861Q) and three main in-frame deletions (delE746-A750, delL747-T751inS, and delL747-P753inS).18 This group also carried out a functional analysis of two of the identified mutations (L858R and delL747-P753insS) and ectopically overexpressed these two mutations and wild-type EGFR in COS-7 cells. In response to the EGFR ligand EGF, Cos-7 cells harboring the mutations had a higher phosphorylation activity of EGFR (assessed by immunoblotting for the EGFR tyrosine-residue Y1068) and were more susceptible to gefitinib as compared with wild-type EGFR-expressing Cos-7 cells,20 corroborating the notion that tumors with these indicated mutations may be more responsive to EGFR TKI treatment.
The distribution of these mutations around the ‘catalytic kinase domain’ is distinct in lung adenocarcinoma and contrasts the mutations in glioblastomas that are located in the extracellular portion of EGFR. Both lead to a constitutively active EGFR in a ligand-independent manner.18 These EGFR TKIs exert marginal effects on glioblastomas harboring EGFR mutations, which are not distributed around the intracellular TK domain.18
A larger study involving 617 NSCLC tumor samples identified mutations in the EGFR gene in 21% of the cases and confirmed the initial findings.21 These mutations were predominantly observed in adenocarcinomas, more common in nonsmokers and in patients from east Asia.21 Separate studies addressed the important issue of whether EGFR TK domain mutations are an early event in the development of lung adenocarcinoma.22 Tang et al22 performed microdissection in 21 tumors with confirmed EGFR mutations, revealing nine of these tumors with the same EGFR mutation in adjacent non-neoplastic alveolar tissue. Based on their findings, they concluded that EGFR mutations are an early event and may represent a target for ‘chemoprevention strategies’. From a mechanistic point of view one would expect to appreciate some morphological change in tissue harboring an EGFR mutation. An explanation for the finding of this group may represent mutations in atypical adenomatous hyperplasia (AAH), a subtle precursor lesion for adenocarcinoma. As these lesions are incidentally found in patients with lung cancer and emphysema, the chemoprevention point may also be valid in this setting. Other groups have identified EGFR mutations in AAH,23 although to varying frequencies. In this context, Yoshida et al23 found EGFR mutation in only 3% of their AAH cases, whereas adenocarcinoma in-situ and invasive adenocarcinoma harbored mutation in 10.8% and 41.9% of cases, respectively. Investigators from the Aichi Cancer Center in Japan found a higher rate of EGFR mutation in AAH of 25%, although their overall EGFR mutation rate in AdCa was also higher.24
From the current body of available literature, three defined regions (exons 18–21) in the EGFR gene are commonly mutated and predict sensitivity to EGFR TKI. These mutations are summarized in Figure 2; the majority are exon 21 point mutations and several in-frame deletions in exon 19.25

Epidermal growth factor receptor (EGFR) mutations and their frequency based on COSMIC annotations. The amino-acid location based upon the exon structure of EGFR is highlighted to show areas of mutation that alter amino-acid sequence. The common mutations are noted to encompass alterations reported in COSMIC. Because many data entries in COSMIC reflect studies focused on exons 21 and 19, the frequency of rarer mutations may be underestimated.
According to Eberhard et al25 and others,18, 26 the most common of these mutations in the EGFR gene involve the point mutation, L858R, in exon 21 and an exon 19 deletion, del746-750. According to Sakurada et al,27 these comprise up to 86% of EGFR mutations in NSCLCs overall. Concerning the sensitivity of these above-mentioned mutations, it has been suggested that in-frame deletions of EGFR predict higher sensitivity to TKI compared with point mutations in exons 18 and 21.28, 29 Specifically, Mitsudomi et al29 reported that lung adenocarcinomas harboring the EGFR exon 19 deletion (del746-750) may be more susceptible to gefitinib as compared with tumors with the exon 21 point mutation (L858R). Additional mutations in the EGFR gene have been identified, in exons 20 and 18. Given the paucity of these rarer mutations, their impact on predicting sensitivity or resistance to EGFR TKI remains to be fully elucidated in larger patient populations (Table 1).25
However, based on current knowledge, some observations have been made regarding rarer mutations and EGFR TKI therapy. The presence of L861Q mutations may be associated with stable disease, but response has not been consistently reported in the three studies containing such cases, with documentation of progressive disease and shorter response duration after EGFR TKI therapy.30, 31, 32 This suggests that EGFR mutation L861Q is not associated with favorable response to EGFR TKI, but admittedly represents an experience of <10 total patients. Although exon 18 mutations are thought to predict response, these too have relatively limited published experience31, 33, 34 with EGFR TKI response in only 2 of 11 patients.
There are three major exon 20 alterations—two are point mutations and the third encompasses a variety of insertion mutations. It has been noted that different exon 20 mutations, also seen in combination with the more frequent exons 21 and 19 mutations, may predict different EGFR TKI response.35 The S768I mutation is reportedly associated with poorer EGFR TKI response;36 and as a single mutation this appears to be so.32, 37 However, double mutations of S768I with L858R or exon 19 deletion have had reports of response again with very few patient cases.30, 37
Exon 20 insertions are reported as a potential cause of TKI resistance.38 This has been supported in subsequent studies,31, 32, 37 albeit with few total patients studied. Although relatively rare in the COSMIC database (possibly because not all recorded studies assay all four EGFR exons), one study showed a 2.2% rate among 1500 tested adenocarcinomas.39 In that study, only five patients received TKI therapy; the two who received single-agent TKI therapy showed no response. These authors postulate that exon 20 insertions alter drug affinity, which they support with computer-assisted 3D modeling.
Although T790M mutations are found in up to 63% of EGFR TKI-treated tumors as a form of acquired resistance (see section ‘Acquired EGFR mutations in lung adenocarcinomas after EGFR receptor inhibitor treatment’), primary resistance due to T790M mutations is thought to be less frequently encountered, with conflicting results using EGFR assays of different sensitivity.40, 41 In rare instances, the T790M mutation is a germline mutation. In a very small subset of patients with NSCLCs (2 patients out of 369 patients, 0.54%), this alteration causes primary resistance to EGFR TKIs.42
To further explore the mechanism of how EGFR inhibitors elicit cell death in lung adenocarcinomas harboring EGFR-sensitizing mutations, several groups hypothesized that gefitinib may elicit apoptotic cell death, depending on the Bcl-2 family of proteins, which is reminiscent of imatinib-mediated cell death induction in CML cells harboring the bcr-abl rearrangement.43 Specifically, they postulated that erlotinib induces apoptosis by utilizing the pro-apoptotic Bcl-2 family of proteins.43 Generally, the Bcl-2 family of proteins consists of members that antagonize the apoptotic cell death, for example, Bcl-2, Bcl-Xl, and Mcl-1, and proteins that drive apoptosis such as Bad, Bax, Puma, and Bim.44 In the context of EGFR inhibition in lung adenocarcinomas with a sensitizing EGFR mutation, erlotinib elicited an upregulation of the pro-apoptotic protein Bim (BCL2L11), which was implicated and instrumental in TKI-induced apoptosis in EGFR-mutated cell lines.43, 45
In addition to apoptosis, other forms of cell death have been described in the context of EGFR inhibition such as autophagy. Autophagy is an intracellular process in which cellular organelles, such as mitochondria, will be engulfed in lysosomal vesicles. It represents a form of cellular stress response to a number of factors or environmental changes, such as hypoxia, withdrawal of growth factor, or drug treatment. Autophagy may exert a pro-survival effect, whereas in other situations it mediates loss of cellular viability. By itself, EGFR-mediated signaling increases the activity mTOR, which in turn exerts an inhibitory effect on autophagy. Thus, inhibition of EGFR signaling could impact autophagy through downstream inhibition of mTOR. A very recent study has linked mutated EGFR with autophagy by demonstrating a molecular interaction between EGFR and Beclin 1, an important mediator of autophagy with tumor-suppressor properties. EGFR-induced Beclin-1 phosphorylation inhibits autophagy, thus driving tumor cell growth and TKI resistance in vivo.46 Thus, this group46 concluded that in the context of TKI-sensitizing EGFR mutations, TKI-induced autophagy enhances cell death. In contrast, a different group linked EGFR TKI treatment combined with autophagy inhibition to cell death.47 Their study was not in the context of activating EGFR mutations. However, this underscores the need for further study to delineate the role of autophagy in TKI treatment of lung adenocarcinoma.
RESISTANCE PATHWAYS
Acquired EGFR Mutations in Lung Adenocarcinomas after EGFR Receptor Inhibitor Treatment
Despite the remarkable response of EGFR mutant lung adenocarcinomas to TKIs, essentially all these tumors recur and eventually develop secondary evolved resistance. These resistant tumors have commonly acquired TKI treatment related EGFR mutations.
One of these mutations is located in exon 20 of the EGFR gene and replaces methionine for a threonine (T790M),48 resulting in increased ATP affinity. This mutation was identified in a patient, who initially responded well to gefitinib treatment (with complete remission) and had an exon 19 mutation in EGFR (delL747–S752).48 After 2 years the tumor recurred, retaining the initially identified exon 19 deletion, but had acquired additionally a second point mutation of the EGFR TK domain in exon 20 (T790M). In a cell culture model, the introduction of the T790M EGFR mutation into Cos-7 cells harboring an EGFR-sensitizing mutation led to resistance of these cells to gefitinib-mediated apoptosis.48 Subsequently, a Japanese study identified the T790M mutation in 7 of 14 patients who had initially responded to EGFR inhibitor treatment.49 Given the fact that mutations in the KRAS gene may confer treatment resistance to EGFR inhibitors, this same group determined whether KRAS mutation might be implicated and contribute to the secondarily evolved EGFR inhibitor resistance.49 Based on their findings, no KRAS mutations were identified in the 14 patients.49 Similar results were found in a study from the United States in which 7 of 16 patients (43%) revealed a T790M mutation.50 In a more recent larger sample set of 155 patients, the T790M mutation was identified in 63% of patients and was by far the most common cause for acquired resistance to EGFR TKIs. Less commonly, they found HER2 (13%) and MET amplification (5%) as a means of resistance.51
EGFR T790M mutations are associated with poorer response and shorter PFS.40, 41 One study raised the possibility that acquired T790M patients had more loco-regional than distant disease failures, and thus had a better post therapy survival,52 but this needs to be corroborated in other series. As the molecular mechanism by which the T790M elicits its resistance to gefitinib appears to be the stronger binding of ATP to the EGFR molecule, irreversible inhibitors of the EGFR kinase activity may still exert activity in tumors with the T790M mutation.
Moreover, lung adenocarcinoma cell lines harboring both a sensitizing EGFR mutation and the T790M mutation exerted an attenuated gefitinib-mediated upregulation of BIM,45 suggesting that BIM is implicated in EGFR inhibitor treatment resistance caused by the T790M mutation. Importantly, this acquired resistance can be overcome by switching to irreversible EGFR inhibitors that have a more sustained effect, and elicit an increase in BIM protein levels, culminating in apoptotic cell death.44 Results of recent clinical trials, investigating irreversible EGFR inhibitors, were completed and published53, 54, 55 Recently, several EGFR inhibitors have been identified in a library screening that specifically target the EGFR T790M mutation, and would in turn counteract the EGFR T790M mutation-driven intrinsic apoptotic resistance of these tumors.56 These molecules were reported to be up to a 100 times more specific for EGFR T790M-mutated tumors compared with wild-type EGFR-expressing tumors.56 Screening lung adenocarcinoma tumor samples for the EGFR T790M mutation is relevant, as these irreversible inhibitors, such as afatinib and dacomitinib, have entered clinical trials.53, 54, 55 For instance, progression-free survival was slightly improved in patients who administered dacomitinib (2.86 month) as compared with erlotinib (1.91 month). However, it is noteworthy that despite the positive impact on progression-free survival, adverse effects were more prominent in patients receiving dacomitinb,53 including skin- and gastrointestinal-related effects.53
Non-mutational pathways of acquired resistance—hepatocyte growth factor/MET
Aside from mutational pathways, activation of survival pathways also contributes to EGFR TKI resistance. Specifically, MET signaling and the amplification of the MET receptor have been shown to hamper the effectiveness of EGFR inhibitors in patients with lung adenocarcinomas.57, 58, 59, 60 MET is a membranous receptor and its expression is increased in lung adenocarcinomas, which is correlated with poorer prognosis.57 This increase in expression is mediated by amplification of the MET gene that, according to the initial study by Engelman et al60, was identified in 22% of patients of a small group of 18 lung adenocarcinomas, which after initial EGFR TKI treatment developed resistance. Thereby, the amplification of the MET gene was confirmed both by FISH and real-time PCR analysis,60 independently validating the amplification status. These findings suggest that MET amplification may be a common event in acquired treatment resistance to EGFR inhibitors in lung adenocarcinoma. Of note, MET amplification is not as common as a primary event, identified in ∼7.3% of EGFR TKI naive cases (Table 2).
Hepatocyte growth factor or ‘scatter factor’61 is the ligand for MET, and upon its binding MET gets auto-phosphorylated, resulting in an activation of several downstream pathways, such as the PI3 kinase pathway, which depends on the receptor protein ERBB3 (HER3).60, 62 Engelman et al60 have linked the HER3 protein to MET amplification-mediated TKI resistance. By performing co-immunoprecipitation, they found that the p85 of subunit of the PI3 kinase readily interacted with phosphorylated HER3 protein, suggesting the downstream involvement of AKT signaling and establishing a ‘MET/ERBB3/PI3K’ signaling axis. In turn, the MET-mediated increase of PI3-kinase signaling rescues the EGFR inhibitor shutdown of PI3 kinase cascade. Recent experimental data also suggests that concomitant inhibition of MET and EGFR signaling in MET-amplified and EGFR-mutated xenografts resulted in an eradication of these tumor xenografts, suggesting that such a combination regimen may also be beneficial in a subset of patients with these molecular alterations.
Histological pattern of acquired resistance—‘small-cell transformation’
Although primary small-cell carcinomas do not harbor EGFR mutations, EGFR mutation-positive adenocarcinomas, when treated with EGFR TKI, can acquire morphological and immunohistochemistry features of small-cell carcinoma. One group described that 5 patients (14%) out of 37 patients in that group with lung adenocarcinoma, harboring a sensitizing EGFR mutations that initially responded to EGFR inhibitor treatment, converted to a small-cell carcinoma that responded to conventional small-cell chemotherapy.63 These five cases did not have T790M mutations or MET amplification; one case developed a detectable PIK3CA mutation. In these cases and in additional case reports, the original EGFR mutation remained detectable in the small-cell carcinoma.64, 65, 66
Non-mutational pathways of acquired resistance, HER2 amplification
As a further mechanism of acquired resistance, HER2 amplification was recently described as a contributing factor in 13% of 155 cases.51 However, according to this study, it could not be excluded that HER2 amplification was present before acquired EGFR TKI resistance.51
Nonresponders to EGFR TK Inhibitor (Table 2)
KRAS mutations in EGFR TK inhibitor nonresponders
Certain mutations of the EGFR downstream signaling confer resistance to EGFR inhibitor treatments. Probably, the most prominent member out of this group is KRAS gene that often exhibits point mutations in codons 12 and 13, located in exon 2.67 The KRAS gene encodes for the GTP-binding protein, RAS. Once mutated, RAS loses its ability to hydrolyze GTP to GDP, becoming constitutively active, disconnects from the upstream EGFR signaling, and thereby becomes independent of the EGF ligand. These mutations appear to be more common in patients with a significant smoking history as compared with patients with EGFR mutations who are more commonly light or nonsmokers.67 KRAS and EGFR mutations generally occur in a mutually exclusive pattern, although some cases have been identified in which both EGFR and KRAS were mutated in the same tumor.68 Despite harboring sensitizing EGFR mutation, patient having mutations in both genes were nevertheless resistant to EGFR inhibitor treatment.68 Therefore, KRAS mutation predicts nonresponse to EGFR TKI either as a result of the mutually exclusive occurrence or as the fact that pathway activation becomes at least partly independent of EGFR activity. However, a recently published phase II clinical trial suggests that EGFR inhibitors in combination with other drugs may even elicit antitumor effects in the presence of KRAS mutations.69 Such a treatment response was achieved by the combination of erlotinib with tivantinib (ARQ 197; ArQule, Woburn, MA, USA), a novel compound that is targeting the MET pathway.69
BRAF mutations and resistance
BRAF is a part of the ERK signaling pathway, and mutations occur in lung adenocarcinomas at a rate of ∼3%.70 Although V600E mutations are the most common in lung adenocarcinoma, a significant proportion of other mutations occur mostly around amino acid 600 but with 469 and 592 as other mutation hot spots. These additional mutational hot spots are seen in lung adenocarcinoma more frequently than in melanoma.71 These are also generally mutually exclusive with EGFR and KRAS mutations, and may also necessitate therapies that are further downstream in the signaling pathway.
Some BRAF mutations are inactivating,72 and these can coexist with EGFR mutations. Such inactivating mutations are located in exon 11 of BRAF, for example, G466V and Y472C. The role of these mutations in the pathobiology of EGFR mutations and EGFR TKI therapy remains to be elucidated.
MEK1 mutations and resistance
The ERK signaling cascade harbors additional kinase downstream of BRAF.73 Although MEK1 mutations may be relatively uncommon in lung adenocarcinoma,74 they may also uncouple the signaling from upstream activations as a mechanism of resistance. Also, MEK targeting may offer a more effective method of pathway inactivation regardless of which upstream kinase is activated.75
The role of PIK3CA mutations in EGFR TKI resistance
The PIK3CA gene is mutated in cases of lung adenocarcinoma at a rate of ∼4%,70 and is found as mostly point mutations altering amino acids 542, 545, and 1047 in roughly two-thirds of cases.76 PIK3CA encodes PI3K that phosphorylates PIP2 to give rise to PIP3, driving activation of AKT and mTOR. Mutations in the PIK3CA gene cause constitutive activation and uncoupling from either ligand-dependent or constitutively active EGFR signaling. This has been shown to confer gefitinib resistance in EGFR-mutated cell lines77 and as a mechanism of acquired resistance in a subset (5%) of treated patients.63 These findings may identify patients requiring combination therapy. Several inhibitors of PI3K have been developed and their use is currently under investigation.
Loss of PTEN as a rare cause of EGFR TKI resistance
Recent data suggests that PTEN is implicated in EGFR TKI-resistant lung cancer. From a mechanistic point of view, loss of PTEN activity causes an EGFR-independent activation of the PIK-3 signaling axis. EGFR phosphorylation may be increased in the absence of PTEN activity.11 The possibility of PTEN loss contributing to resistance was suggested by the finding of one case of PTEN deletion in 24 EGFR-mutated lung adenocarcinoma.11 Observation of PTEN deletion in unexpected primary resistance or acquired resistance remains to be well documented, and more clinical experience is needed to define the role of PTEN in EGFR TKI resistance pathobiology.
HER2 mutations
Mutations in ERBB2 are found in ∼2% of lung adenocarcinomas, and the most frequent alteration is an in-frame duplication/insertion at codons 776–779.21, 78 These are also generally mutually exclusive with EGFR and KRAS mutations.
Germline mutation in BIM
Recent research has shown that BCL2L11, encoding for BIM, has a pivotal role in TKI-mediated cell death in lung adenocarcinoma and CML. In this context, a germline mutation (deletion or polymorphism) in the BCL2L11 gene led to an alternative spliced variant of BIM that is devoid of the apoptosis mediating ‘pro-apoptotic BCL-2 homology domain 3’.79 This inherited alternative BIM variant conferred less responsiveness to TKI in patients with lung adenocarcinomas.79 Ironically, akin to the epidemiological distribution pattern of EGFR mutations in general, there was a preponderance of BIM germline mutations in patients from East Asia.80 In fact, in a group of 2597 patients, the mutation was detected in 12.3% of East Asia and absent in patients with African or European descent.80 Patients with a BIM polymorphism revealed a 6.6-month median progression-free survival compared with 11.9 months in patients without it.79 Drugs that inhibit apoptosis inhibtors—BCL-2 homology domain 3 mimetics or Smac mimetics—drive intrinsic apoptotic signaling and may find use in conjunction with EGFR-targeting TKI therapy.80
Translocations—anaplastic lymphoma kinase, ROS1, and RET
Anaplastic lymphoma kinase translocations as a 2p inversion resulting in a fusion gene with echinoderm microtubule-associated protein-like 4 have been described in a subset of lung adenocarcinoma, representing between 2 and 5% of these tumors overall.81 In addition, this alteration was not seen in tumors harboring EGFR or KRAS mutations.82 Specifically, while this alteration is associated with sensitivity to crizotinib,83 it is not associated with response to EGFR-targeting TKI.84
Less frequently encountered than translocations involving anaplastic lymphoma kinase, translocations involving RET and ROS have been identified. Translocations involving ROS1 have been described with CD74 and SLC34A2, and may confer sensitivity to crizotinib.85 Although rare, this alteration is also seen in EGFR wild-type tumors and predicts nonresponse to EGFR TKI therapy.86 Translocations involving RET and KIF5b, and RET with CCDC6 have been reported in lung adenocarcinomas (∼1%), also mutually exclusive with EGFR mutations;87 while therapeutic data is sparse, the mutual exclusivity suggests that this alteration is also associated with primary resistance to EGFR TKI.
CONCLUDING REMARKS
Over the last decade, significant advancements have been made in our understanding of the pathobiology of lung adenocarcinoma. The advances in the understanding of EGFR-activating mutations in lung adenocarcinoma has made this one of the model paradigms in targeted therapy. Identification of associated or acquired molecular events may require addition of new agents, including combinations of drugs with different molecular targets. The relatively low frequency of some of these events represent a challenge toward developing cohort sizes sufficient for the practice of evidence-based medicine. Nevertheless, further understanding of the complex interplay of members of this pathway, activation of parallel pathways, and acquisition of secondary mutational events continues to uncover important issues in the continued management decisions in lung adenocarcinoma patients.
References
Howlader NNA, Krapcho M, Garshell J et alSEER Cancer Statistics Review, 1975-2010. In: Institute. NC, ed. Bethesda, MD2013.
da Cunha Santos G, Shepherd FA, Tsao MS . EGFR mutations and lung cancer. Ann Rev Pathol 2011;6:49–69.
Hochhaus A . Imatinib mesylate (Glivec, Gleevec) in the treatment of chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors (GIST). Ann Hematol 2004;83 (Suppl 1):S65–S66.
Khazaie K, Schirrmacher V, Lichtner RB . EGF receptor in neoplasia and metastasis. Cancer Metastasis Rev 1993;12:255–274.
Ullrich A, Schlessinger J . Signal transduction by receptors with tyrosine kinase activity. Cell 1990;61:203–212.
Carpenter G . Receptors for epidermal growth factor and other polypeptide mitogens. Ann Rev Biochem 1987;56:881–914.
Libermann TA, Nusbaum HR, Razon N et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985;313:144–147.
Huang HS, Nagane M, Klingbeil CK et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997;272:2927–2935.
Liebmann C . Regulation of MAP kinase activity by peptide receptor signalling pathway: paradigms of multiplicity. Cell Signal 2001;13:777–785.
Jorissen RN, Walker F, Pouliot N et al. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res 2003;284:31–53.
Sos ML, Koker M, Weir BA et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009;69:3256–3261.
Guo Y, Du J, Kwiatkowski DJ . Molecular dissection of AKT activation in lung cancer cell lines. Mol Cancer Res 2013;11:282–293.
Takata S, Takigawa N, Segawa Y et al. STAT3 expression in activating EGFR-driven adenocarcinoma of the lung. Lung Cancer 2012;75:24–29.
Weinstein IB, Joe AK . Mechanisms of disease: oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 2006;3:448–457.
Fukuoka M, Yano S, Giaccone G et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003;21:2237–2246.
Kris MG, Natale RB, Herbst RS et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003;290:2149–2158.
Shepherd FA, Rodrigues Pereira J, Ciuleanu T et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123–132.
Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129–2139.
Pao W, Miller V, Zakowski M et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA 2004;101:13306–13311.
Yarden Y, Sliwkowski MX . Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–137.
Shigematsu H, Lin L, Takahashi T et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl Cancer Inst 2005;97:339–346.
Tang X, Shigematsu H, Bekele BN et al. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005;65:7568–7572.
Yoshida Y, Shibata T, Kokubu A et al. Mutations of the epidermal growth factor receptor gene in atypical adenomatous hyperplasia and bronchioloalveolar carcinoma of the lung. Lung Cancer 2005;50:1–8.
Sakamoto H, Shimizu J, Horio Y et al. Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasive to invasive adenocarcinomas. J Pathol 2007;212:287–294.
Eberhard DA, Johnson BE, Amler LC et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005;23:5900–5909.
Paez JG, Janne PA, Lee JC et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497–1500.
Sakurada A, Shepherd FA, Tsao MS . Epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer: impact of primary or secondary mutations. Clin Lung Cancer 2006;7 (Suppl 4):S138–S144.
Chou TY, Chiu CH, Li LH et al. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin Cancer Res 2005;11:3750–3757.
Mitsudomi T, Kosaka T, Endoh H et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol 2005;23:2513–2520.
Yang CH, Yu CJ, Shih JY et al. Specific EGFR mutations predict treatment outcome of stage IIIB/IV patients with chemotherapy-naive non-small-cell lung cancer receiving first-line gefitinib monotherapy. J Clin Oncol 2008;26:2745–2753.
Locatelli-Sanchez M, Couraud S, Arpin D et al. Routine EGFR molecular analysis in non-small-cell lung cancer patients is feasible: exons 18-21 sequencing results of 753 patients and subsequent clinical outcomes. Lung 2013;191:491–499.
Lee VH, Tin VP, Choy TS et al. Association of Exon 19 and 21 EGFR mutation patterns with treatment outcome after first-line tyrosine kinase inhibitor in metastatic non-small-cell lung cancer. J Thorac Oncol 2013;8:1148–1155.
Han SW, Kim TY, Hwang PG et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol 2005;23:2493–2501.
Takano T, Ohe Y, Sakamoto H et al. Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancer. J Clin Oncol 2005;23:6829–6837.
Wu JY, Wu SG, Yang CH et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res 2008;14:4877–4882.
Asahina H, Yamazaki K, Kinoshita I et al. Non-responsiveness to gefitinib in a patient with lung adenocarcinoma having rare EGFR mutations S768I and V769L. Lung Cancer 2006;54:419–422.
Lund-Iversen M, Kleinberg L, Fjellbirkeland L et al. Clinicopathological characteristics of 11 NSCLC patients with EGFR-exon 20 mutations. J Thorac Oncol 2012;7:1471–1473.
Greulich H, Chen TH, Feng W et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2005;2:e313.
Arcila ME, Nafa K, Chaft JE et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013;12:220–229.
Rosell R, Molina MA, Costa C et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res 2011;17:1160–1168.
Su KY, Chen HY, Li KC et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non-small-cell lung cancer. J Clin Oncol 2012;30:433–440.
Girard N, Lou E, Azzoli CG et al. Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: a preliminary report. Clin Cancer Res 2010;16:755–763.
Gong Y, Somwar R, Politi K et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med 2007;4:e294.
Siegelin MD . Inhibition of the mitochondrial Hsp90 chaperone network: a novel, efficient treatment strategy for cancer? Cancer Lett 2013;333:133–146.
Costa DB, Halmos B, Kumar A et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 2007;4:1669–1679 discussion 80.
Wei Y, Zou Z, Becker N et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013;154:1269–1284.
Han W, Pan H, Chen Y et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PloS ONE 2011;6:e18691.
Kobayashi S, Boggon TJ, Dayaram T et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005;352:786–792.
Kosaka T, Yatabe Y, Endoh H et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006;12:5764–5769.
Balak MN, Gong Y, Riely GJ et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006;12:6494–6501.
Yu HA, Arcila ME, Rekhtman N et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240–2247.
Oxnard GR, Arcila ME, Sima CS et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res 2011;17:1616–1622.
Ramalingam SS, Blackhall F, Krzakowski M et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2012;30:3337–3344.
Miller VA, Hirsh V, Cadranel J et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012;13:528–538.
Yang JC, Shih JY, Su WC et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol 2012;13:539–548.
Zhou W, Ercan D, Chen L et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009;462:1070–1074.
Turke AB, Zejnullahu K, Wu YL et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010;17:77–88.
Yano S, Wang W, Li Q et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008;68:9479–9487.
Cheng TL, Chang MY, Huang SY et al. Overexpression of circulating c-met messenger RNA is significantly correlated with nodal stage and early recurrence in non-small cell lung cancer. Chest 2005;128:1453–1460.
Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039–1043.
Bottaro DP, Rubin JS, Faletto DL et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251:802–804.
Graziani A, Gramaglia D, Cantley LC et al. The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J Biol Chem 1991;266:22087–22090.
Sequist LV, Waltman BA, Dias-Santagata D et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3 75ra26.
Watanabe S, Sone T, Matsui T et al. Transformation to small-cell lung cancer following treatment with EGFR tyrosine kinase inhibitors in a patient with lung adenocarcinoma. Lung Cancer 2013;82:370–372.
Popat S, Wotherspoon A, Nutting CM et al. Transformation to ‘high grade’ neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer 2013;80:1–4.
Alam N, Gustafson KS, Ladanyi M et al. Small-cell carcinoma with an epidermal growth factor receptor mutation in a never-smoker with gefitinib-responsive adenocarcinoma of the lung. Clinical Lung Cancer 2010;11:E1–E4.
Pao W, Wang TY, Riely GJ et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005;2:e17.
Takeda M, Okamoto I, Fujita Y et al. De novo resistance to epidermal growth factor receptor-tyrosine kinase inhibitors in EGFR mutation-positive patients with non-small cell lung cancer. J Thorac Oncol 2010;5:399–400.
Sequist LV, von Pawel J, Garmey EG et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol 2011;29:3307–3315.
Bamford S, Dawson E, Forbes S et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004;91:355–358.
Paik PK, Arcila ME, Fara M et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–2051.
Cardarella S, Ogino A, Nishino M et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res 2013;19:4532–4540.
Pao W, Girard N . New driver mutations in non-small-cell lung cancer. Lancet Oncol 2011;12:175–180.
Marks JL, Gong Y, Chitale D et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res 2008;68:5524–5528.
Cortot AB, Repellin CE, Shimamura T et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res 2013;73:834–843.
Kang S, Bader AG, Vogt PK . Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA 2005;102:802–807.
Engelman JA, Mukohara T, Zejnullahu K et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 2006;116:2695–2706.
Buttitta F, Barassi F, Fresu G et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer 2006;119:2586–2591.
Ng KP, Hillmer AM, Chuah CT et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 2012;18:521–528.
Cheng EH, Sawyers CL . In cancer drug resistance, germline matters too. Nat Med 2012;18:494–496.
Soda M, Choi YL, Enomoto M et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561–566.
Gainor JF, Varghese AM, Ou SH et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 2013;19:4273–4281.
Kwak EL, Bang YJ, Camidge DR et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693–1703.
Shaw AT, Yeap BY, Mino-Kenudson M et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009;27:4247–4253.
Bergethon K, Shaw AT, Ou SH et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol 2012;30:863–870.
Kim HR, Lim SM, Kim HJ et al. The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann Oncol 2013;24:2364–2370.
Suehara Y, Arcila M, Wang L et al. Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clin Cancer Res 2012;18:6599–6608.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Siegelin, M., Borczuk, A. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab Invest 94, 129–137 (2014). https://doi.org/10.1038/labinvest.2013.147
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2013.147
Keywords
This article is cited by
-
Integrative multi-omics characterization reveals sex differences in glioblastoma
Biology of Sex Differences (2024)
-
EGFR phosphorylates HDAC1 to regulate its expression and anti-apoptotic function
Cell Death & Disease (2021)
-
Radiomics signature of brain metastasis: prediction of EGFR mutation status
European Radiology (2021)
-
Overexpression of BZW1 is an independent poor prognosis marker and its down-regulation suppresses lung adenocarcinoma metastasis
Scientific Reports (2019)
-
A Nexus model of cellular transition in cancer
Biological Research (2018)
